Metallocene
Metallocene sind eine Gruppe von metallorganischen Verbindungen, in denen ein zentrales Metallatom wie in einem Sandwich zwischen zwei Cyclopentadienyl-Liganden (C5H5, Abkürzung: Cp) angeordnet ist. Entsprechend werden Metallocene als Sandwichverbindungen bezeichnet. Die Entdeckung des ersten Metallocens, Ferrocen, das 1951 unabhängig von zwei verschiedenen Forschungsteams durch Zufall synthetisiert wurde, gilt heute als einer der Meilensteine in der metallorganischen Chemie und war der Startpunkt für eine rasante Entwicklung einer neuen Sparte der Chemie. Über 80 % der metallorganischen Komplexe sind heute Cyclopentadienyl-Komplexe.[1]


Metallocene finden vielfache Anwendung. Sie werden für die Herstellung von Leuchtdioden eingesetzt. Derivate der Metallocene wie Titanocendichlorid eignen sich als Katalysatoren für die Olefin-Polymerisation mit hoher Produktivität und Selektivität. In der Tumortherapie zeigen einige Metallocenderivate zytostatische Eigenschaften. Ferrocen kann unter anderem dazu verwendet werden, die Klopffestigkeit von Treibstoffen (als Ersatz von Bleitetraethyl) zu erhöhen und deren Verbrennung zu verbessern.
1973 erhielten Ernst Otto Fischer und Geoffrey Wilkinson für ihre Arbeiten über metallorganische Verbindungen und die Aufklärung der Bindungsverhältnisse im Ferrocen den Nobelpreis für Chemie.[2]
Geschichte
Bearbeiten1951 versuchten Tom J. Kealy und Peter Pauson an der Duquesne University die Darstellung von Fulvalen durch die Reaktion von Eisen(III)-chlorid mit Cyclopentadienylmagnesiumbromid entsprechend der folgenden Gleichung durchzuführen. Statt des erwarteten Fulvalens erhielten Kealy und Pauson als Hauptprodukt orangefarbene Kristalle, die überraschenderweise an der Luft und bei Temperaturen über 300 °C stabil waren und leicht sublimiert werden konnten.[3]

Die erste Elementaranalyse für Kohlenstoff und Wasserstoff zeigte, dass es sich bei der Verbindung nicht um Fulvalen handeln konnte, und nach einigen Berechnungen wurde die Summenformel C10H10Fe vorgeschlagen. Die qualitative und quantitative Analyse des Eisens gestaltete sich jedoch schwierig, da die Verbindung selbst gegenüber konzentrierter Schwefelsäure beständig war. Erst durch Kochen mit konzentrierter Salpetersäure gelang der qualitative Eisennachweis. Zur quantitativen Analyse musste die Substanz sogar mit Perchlorsäure (HClO4) bis zur Trocknung abgeraucht werden.[4]
Völlig unabhängig – und zeitlich vor der Arbeit von Tom J. Kealy und Peter L. Pauson – hatten im gleichen Jahr Samuel A. Miller, John A. Tebboth und John F. Tremaine bei der British Oxygen Company die gleiche Substanz durch die Reaktion von Cyclopentadien-Dampf mit frisch reduziertem Eisen bei 300 °C hergestellt und beschrieben.[5][4]


Miller und seine Kollegen reichten ihre Arbeit sogar vor Kealy und Pauson zur Veröffentlichung ein, aber da Nature, die Fachzeitschrift, in der Kealy und Pauson veröffentlichten, schneller publizierte, wurde deren Beitrag eher veröffentlicht als der von Miller. Laut Miller hatte dieser die Substanz bereits drei Jahre zuvor synthetisiert.[6] Möglicherweise wurde das Ferrocen sogar noch einige Jahre früher bei Union Carbide, welche Versuche zur katalytischen Spaltung von Cyclopentadien in Eisenrohren durchführten, zum ersten Mal hergestellt.[4] Dort wurde es jedoch nie weiter beachtet oder analysiert.

Obwohl die Stabilität der Substanz auf eine andere Art der Bindung als in den wenigen bis dahin bekannten metallorganischen Verbindungen, wie etwa dem Zeise-Salz K[PtCl3(C2H2)], schließen ließ, gingen Kealy und Pauson zunächst von einer rein kovalenten Bindung des Cyclopentadienylringes mit dem Eisen aus und schlugen die nebenstehende Struktur vor. Die Gruppe um Miller ging dagegen von einer eher ionischen Struktur aus. Aufgrund der Infrarotdaten (nur eine C-H-Schwingung, entsprechend nur eine Art der C-H-Bindung im Cp-Ring) und des gefundenen Diamagnetismus schlossen 1952 Geoffrey Wilkinson und Robert B. Woodward an der Harvard University auf eine Art Sandwichstruktur.[7] Noch im gleichen Jahr bestätigten Ernst Otto Fischer und Wolfgang Pfab in München, sowie Philip Frank Eiland und Ray Pepinsky am Pennsylvania State College diese Struktur mittels Röntgen-Kristallstrukturanalyse.[8][9][10] Als Woodward postulierte, dass die Cyclopentadienylringe im Fe(C5H5)2 einer elektrophilen Substitution zugänglich sein sollten, führten Whitning und Rosenblum die erste Friedel-Crafts-Acylierung am Cyclopentadienylring im Ferrocen durch.[11] Durch dieses für aromatische Substanzen typische Verhalten kam, in Analogie zur englischen Endung -ene für aromatische Substanzen (z. B. Benzene für Benzol), der Namensvorschlag Ferrocen zustande.

Fast in einer Art Wettstreit synthetisierten die beiden Arbeitsgruppen um Ernst Otto Fischer in München und Geoffrey Wilkinson in Harvard in den folgenden Jahren in schneller Folge eine Vielzahl Biscyclopentadienyl-Komplexe anderer Übergangsmetalle, sowie deren Derivate:
- 1952: Titanocendibromid, Zirconocendibromid, Vanadocendichlorid, Ruthenocen und das Ruthenocenium-Kation[6][12][13]
- 1953: Nickelocen,[14] Cobaltocen,[15] Nickelocenium-Kation, Chromocen, Rhodocenium-Kation und Iridocenium-Kation[16][17][18]
- 1954: Vanadocen,[19] Titanocenhydroxybromid, Magnesocen, Manganocen, Niobocentribromid, Tantalocentribromid und Rhenocenhydrid[6][20][21][22]
In den Jahren 1954 und 1955 wurden die Tricyclopentadienyl-Komplexe von Scandium, Yttrium, Lanthan und der Lanthanoide Cer, Praseodym, Neodym, Samarium, Dysprosium, Erbium und Ytterbium synthetisiert und beschrieben. Für diese wurde, aufgrund einer raschen und vollständigen Reaktion mit Eisenchlorid zu Ferrocen, eine ionische Struktur vorgeschlagen.[23][24]
Ab 1954 synthetisierten die beiden Arbeitsgruppen sogenannte Halbsandwichkomplexe, welche nur einen Cyclopentadienyl-Liganden enthalten, wie etwa (C5H5)V(CO)4, (C5H5)Mn(CO)3, (C5H5)Co(CO)2, (C5H5)Ni(CO), (C5H5)Mo(CO)3H, [(C5H5)Fe(CO)2]2 und (C5H5)Fe(CO)2Cl.[6][25] Auch über die durch Carbonyl-Liganden (CO) verbrückten Halbsandwich-Komplexe (C5H5)Mo(CO)6Mo(C5H5) und (C5H5)W(CO)6W(C5H5) wurde berichtet.[18] Diese Halbsandwich-Verbindungen gehören, im Sinne der Definition, streng genommen aber nicht zu den Metallocenen. Neben Metallocenen mit (unsubstituierten) Cyclopentadienyl-Liganden wurden bis heute eine große Zahl von Komplexen mit substituierten Ringen hergestellt. Von großem Interesse ist insbesondere der Pentamethylcyclopentadienyl-Ligand (Abkürzung: Cp*), weil er durch seinen großen Raumanspruch instabile Metallocene sterisch stabilisieren und dadurch isolierbar machen kann.
Herstellung
BearbeitenDa Cyclopentadien bei Raumtemperatur in einer Diels-Alder-Reaktion zu Dicyclopentadien dimerisiert, muss dieses im ersten Schritt durch thermische Spaltung erst wieder in Cyclopentadien überführt werden (Retro-Diels-Alder-Reaktion). In der Praxis wird das Cyclopentadien in Gegenwart eines Katalysators, beispielsweise Eisenpulver, aus seinem Dimer abdestilliert.

Metallocene und allgemein Cyclopentadienyl-Metall-Verbindungen können auf verschiedenen Wegen hergestellt werden.
Durch Metathese: Die ersten Metallocene wurden mit einem Grignard-Reagenz hergestellt, später wurde stattdessen bevorzugt das Cyclopentadienylnatrium eingesetzt:[26]


Dabei können auch Metalle in der Oxidationsstufe +III eingesetzt werden, welche im ersten Teilschritt zunächst reduziert werden, wobei 9,10-Dihydrofulvalen als Nebenprodukt entstehen kann:

Aber auch die Verwendung von metallischen Reduktionsmitteln wie Zink wird beschrieben:[27]
![{\displaystyle \mathrm {RuCl_{3}(H_{2}O)} _{x}+\mathrm {3\,C_{5}H_{6}\ {\xrightarrow[{-ZnCl_{2},\ -xH_{2}O}]{+Zn,\ Ethanol}}\ Ru(C_{5}H_{5})_{2}+C_{5}H_{8}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/4cd2eea375b38e37e3d368938e4feab78546a94d)
Aufgrund der geringen Stabilität des Magnesocens ist dieses gut zur Cyclopentadienylierung, für die Übertragung von Cp-Einheiten auf andere Metalle, geeignet. Triebkraft ist die Bildung des stabilen Magnesiumdihalogenids.[28]

Allgemein lassen sich Cyclopentadienylalkali-Verbindungen durch Reaktion von 2,4-Cyclopentadienbrom mit Alkaliorganylen herstellen:[29]

Daraus kann dann im nächsten Schritt der Sandwichkomplex Lithocen, der nur in Komplexen als anionisches Lithoceniumions existiert, z. B. durch Reaktion von LiCp mit Tetraphenylphosphoniumchlorid (PPh4Cl) synthetisiert werden:[29]

Durch Disproportionierung: Magnesocen kann direkt aus CpMgBr gewonnen werden. Dazu wird zunächst Cyclopentadienylmagnesiumbromid durch Umsetzung von Ethylmagnesiumbromid mit Cyclopentadien hergestellt, welches dann bei 220 °C und 10−4 mbar zu MgCp2 und MgBr2 disproportioniert (Schlenk-Gleichgewicht):[28]
![{\displaystyle \mathrm {Mg(C_{2}H_{5})Br\;{\xrightarrow[{-C_{2}H_{6}}]{+C_{5}H_{6},\ Et_{2}O}}\;Mg(C_{5}H_{5})Br\ \xrightarrow {220\,^{\circ }C,\,10^{-4}\,mbar} } }](https://wikimedia.org/api/rest_v1/media/math/render/svg/b46e7c28f9053c565a121f53463b20f18a396192)


Aus den Bestandteilen in Gegenwart von Basen: Im Jahre 1954 versuchte Wilkinson Biscyclopentadienyl-Metall-Komplexe mit Hilfe von Aminen als Halogenwasserstoffakzeptoren zu synthetisieren. Allerdings waren die Ausbeuten mit 3–4 % nur sehr gering. Nur beim Ferrocen oder durch die Verwendung stärkerer Basen, wie Kaliumhydroxid lässt sich die Ausbeute deutlich verbessern:[25]

Direkt aus den Bestandteilen: Bei reaktiven Metallen, wie die Alkalimetalle und Erdalkalimetalle können die Cyclopentadienylmetall-Verbindungen direkt durch die Umsetzung des Metalls mit Cyclopentadien hergestellt werden:[30]

![{\displaystyle \mathrm {Mg+2\ C_{5}H_{6}\;{\xrightarrow[{}]{500\,^{\circ }C}}\;Mg(C_{5}H_{5})_{2}+H_{2}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/f4a0ee497ecdd911dc5c3083247c43addaca7f94)
![{\displaystyle \mathrm {M+2\ C_{5}H_{6}\;{\xrightarrow[{2.Vakuumsubl.\,260\,^{\circ }C\,-\,360\,^{\circ }C}]{1.THF/DMF,-H_{2}}}\;M(C_{5}H_{5})_{2}\;:M=Ca,Sr} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/efe80c0e43a8bc3227f3d91df846aa7434d69767)
Bei den Elementen der Gruppe 4 bis 12 gelingt dies nur bei dem besonders stabilen Ferrocen.[29][5]
![{\displaystyle \mathrm {2\ C_{5}H_{6}+Fe\;{\xrightarrow[{}]{300\,^{\circ }C}}\;Fe(C_{5}H_{5})_{2}+H_{2}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/abba1f5ec64302fd28a74d2c42deca8983871a50)
Ferrocen und Cobaltocen können zudem bei Verwendung von Eisen- oder Cobalt-Anoden und Tetrabutylammoniumbromid (Bu4NBr) als Leitsalz direkt in einer elektrochemischen Reaktion erzeugt werden:[31]
![{\displaystyle \mathrm {M+2\ C_{5}H_{6}\;{\xrightarrow[{}]{Bu_{4}NBr,\,20\,^{\circ }C}}\;M(C_{5}H_{5})_{2}+H_{2}\;:M=Fe,Co} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/fe3daacec2b4d57d27a93445fa4d7e4342679f50)
Aus Metallhydriden oder Metallorganylen: Die Biscyclopentadienyl-Komplexe von Calcium, Strontium und Barium können durch Umsetzung der entsprechenden Hydride mit Cyclopentadien gewonnen werden:[30]
![{\displaystyle \mathrm {MH_{2}+2\ C_{5}H_{6}\;{\xrightarrow[{}]{260\ ^{\circ }C\ -\ 400\ ^{\circ }C}}\;M(C_{5}H_{5})_{2}+H_{2}\;:M=M=Ca,Sr,Ba} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/60ec23220197727bceff979746a08c7d69250095)
Für den Labormaßstab ist die Umsetzung von Diorganylmetall-Verbindungen, wie Dibutylmagnesium, mit Cyclopentadien möglich.[32]

Die Metallocene der Gruppe 4 und 5 lassen sich durch Reduktion der entsprechenden Metallocendichloride mit metallischen Natrium herstellen:[33]

Nomenklatur, Struktur und Haptizität
Bearbeiten
In Analogie zum ersten Vertreter dieser Substanzklasse, dem Ferrocen, bezeichnet man als Metallocene im engeren Sinne nach IUPAC nur die Bis(cyclopentadienyl)metall-Komplexe der Nebengruppenelemente, in denen die Cyclopentadienylringe koplanar angeordnet sind und so mit dem Metallatom eine klassische Sandwichstruktur bilden (s. u.).[34]
Die Cyclopentadienyl-Liganden können aber, je nach Komplex und Zentralatom, auf verschiedene Art gebunden sein – dies bezeichnet man als Haptizität, dargestellt durch den griechischen Kleinbuchstaben η: in einer klassischen Sandwichstruktur weisen beide Cyclopentadienylringe eine η5 (pentahapto) Koordination auf; dies entspricht der am häufigsten auftretenden gleichmäßigen Bindung des zentralen Metallatoms über alle 5 Kohlenstoffatome und gleichen Metall-Kohlenstoff-Abständen. Bei einer η1 (monohapto) Koordination wird nur ein Ringatom für die Bindung benutzt. Die anderen Strukturelemente mit η2 (dihapto), η3 (trihapto) und η4 (tetrahapto) existieren ebenfalls, sind aber sehr viel seltener.[35] Als Metallocene im weiteren Sinne bezeichnet man alle Verbindungen des Typs MCp2, auch dimere oder polymere Komplexe, wie etwa Titanocen oder Manganocen oder auch die Komplexe der Hauptgruppenelemente, die von der Sandwichstruktur abweichen.[36]


Die klassische Sandwichstruktur tritt bei den Metallocenen der ersten Reihe der Übergangsmetalle von Vanadium bis Nickel, der Eisengruppe (Eisen, Ruthenium, Osmium) sowie wenigen anderen Metallen, inklusive Hauptgruppenelementen auf.[37] η5,η5-Sandwichkomplexe können in zwei Konformationen, ekliptisch (auf Deckung) und gestaffelt (auf Lücke), auftreten. Die Rotationsbarriere zwischen den beiden Konformeren ist bei unsubstituierten Cp-Ringen nur sehr klein; die Aktivierungsenergie beträgt bei Ferrocen, Ruthenocen und Osmocen zwischen 8 und 21 kJ·mol−1.[37]
Die Metallocene der frühen Übergangsmetalle tendieren dazu, keine klassische Sandwichstruktur zu bilden, da diese Elektronenmangelverbindungen sind (sie haben deutlich weniger als 18 Valenzelektronen). Durch Abwinkelung der Cp-Ringe auf einen Winkel von 130° kann das zentrale Metallatom bis zu drei weitere Liganden tragen, wodurch sich die Anzahl der Valenzelektronen erhöht.[37] Ein bekannter Vertreter ist das Schwartz-Reagenz (s. u.), in der das Zirconiumatom von insgesamt vier Liganden umgeben ist. Durch die Verwendung des elektronenreicheren und sterisch anspruchsvolleren Liganden Pentamethylcyclopentadienyl (Cp*) ist es möglich, diese reaktiven (weil elektronenarmen) Komplexe in der klassischen Sandwichstruktur zu stabilisieren.


Werden in Metallocenen die Cyclopentadienylringe durch Kohlenwasserstoffbrücken verbunden, ergibt sich die Verbindungsklasse der Metallocenophane, von denen der erste Vertreter, das [4]Ferrocenophan, 1958 von Arthur Lüttringhaus u. a. synthetisiert wurde.[38][39]
Im weiteren Sinn werden unter dem Begriff Metallocenverbindungen auch Halbsandwich-Verbindungen verstanden, die nur ein Cyclopentadienyl-System über π-Bindungen an ein zentrales Metallatom gebunden besitzen. Die Absättigung freier (Elektronen-)Valenzen erfolgt meist über Carbonylgruppen (z. B. Tricarbonyl(η5-cyclopentadienyl)mangan).
Zur Verbindungsklasse der Metallocene zählen manche Lehrbücher ebenfalls das Uranocen, obwohl es sich hierbei nicht um einen Bis(cyclopentadienyl)-, sondern um einen Bis(cyclooctatetraenyl)-Komplex des Urans handelt.
Neben den Metallocenen, die ein Metallatom enthalten, gibt es auch Verbindungen, die zwei Metallatome tragen. Ein solches Dimetallocen wurde mit dem Decamethyldizinkocen erstmals 2004 beschrieben.[40] Ein weiteres Beispiel ist das Diberyllocen.[41] Das erste heterobimetallische Dimetallocen wurde 2024 beschrieben und enthält Lithium und Aluminium als Metalle.[42]
Bindungsmodelle
Bearbeiten18-Elektronen-Regel
BearbeitenDie 18-Elektronen-Regel für die Übergangsmetallelemente ist das Äquivalent zur Oktettregel der Hauptgruppenelemente und kann dazu herangezogen werden, die Stabilität von organometallischen Verbindungen vorherzusagen. Sie besagt, dass organometallische Moleküle oder Komplexe, in denen die Summe der Valenzelektronen des Metalls plus die Bindungselektronen, welche von den Liganden beigesteuert werden, insgesamt 18 beträgt, besonders stabil sind. Beim Fe(η5-C5H5)2 ergibt sich die Anzahl der Valenzelektronen zu


Durch die 18-Elektronen-Regel können sowohl die hohe Stabilität des Ferrocens[3] als auch die des Cobaltocenium- und des Rhodocenium-Kations gut erklärt werden[43] – alle drei Moleküle sind zueinander isoelektronisch und haben 18 Valenzelektronen. Auch kann die Reaktivität von Rhodocen und Cobaltocen erklärt werden. Beide Komplexe haben 19 Valenzelektronen, was dazu führt, dass sie leicht oxidiert werden können und Rhodocen z. B. nur sehr schwierig aus einer Rhodocenium-Lösung isoliert werden kann.[17] Anders als die Stabilität können die Bindungsverhältnisse und die Strukturen in metallorganischen Komplexen durch die 18-Elektronen-Regel nicht erklärt werden.
Kristallfeldtheorie und Ligandenfeldtheorie
BearbeitenDie Kristallfeldtheorie liefert ein qualitatives Verständnis und die Ligandenfeldtheorie erlaubt quantitative Voraussagen über die Eigenschaften von Übergangsmetallsalzen oder -komplexen. Beide Theorien erklären Struktur, Farbe und Magnetismus dieser Substanzen. Beide Theorien betrachten dabei, wie die d-Orbitale des Komplexzentrums durch die Liganden energetisch beeinflusst werden. In einem unkomplexierten Zentralatom sind alle d-Orbitale energetisch entartet, d. h., sie besitzen alle die gleiche Energie. Je stärker ein Ligand mit einem d-Orbital in Wechselwirkung tritt, desto stärker wird dieses energetisch destabilisiert (angehoben), was dann zu einer energetischen Aufspaltung der d-Orbitale führt. Bei den Metallocenen kommt es zu einer 2-1-2 Aufspaltung: die Orbitale in der xy-Ebene (dxy und dx2-y2) treten kaum in Wechselwirkung mit den Cp-Liganden und sind daher energetisch begünstigt. Das dz2 Orbital tritt nur mit einem Teil in Wechselwirkung und liegt in der Mitte. Am stärksten destabilisiert werden die Orbitale dxz und dyz, die vollständig zu den Ringen zeigen.
Molekülorbitaltheorie
Bearbeiten
Weder die 18-Elektronen-Regel noch die Kristallfeldtheorie für sich allein können die Eigenschaften des Ferrocens im vollen Umfang erklären. Erst durch die Entwicklung der Molekülorbitaltheorie (MO-Theorie) war es möglich, Struktur und Stabilität des Ferrocens in einem Modell zu erklären.[44]
Bei der MO-Theorie werden, ebenso wie bei der Kristall- bzw. Ligandenfeldtheorie, Wechselwirkungen der Metall-Orbitale mit den Ligand-Orbitalen betrachtet. Das Ergebnis ist ein Molekülorbital-Diagramm, welches bindende, nicht-bindende und antibindende Orbitale enthält. Wie in der Kristallfeldtheorie resultiert die energetische Aufspaltung der Orbitale dabei aus der Wechselwirkung zwischen Metall- und Ligand-Orbitalen. Treten beispielsweise ein Ligand-Orbital des Cp-Ringes und ein d-Orbital des Metalls in Wechselwirkung, entstehen daraus zwei neue Molekülorbitale (MOs), welche energetisch in ein bindendes und ein antibindendes MO aufgespalten werden. Die Stärke der Aufspaltung (die energetische Anhebung des einen und Absenkung des anderen MOs) ist dabei umso größer, je stärker die Wechselwirkung (räumliche Überlappung) zwischen Ligand- und Metall-Orbital ist. Findet keine Wechselwirkung statt, ändert sich das entsprechende Orbital energetisch nicht und es ergibt sich ein nichtbindendes Orbital. Auch bei der MO-Theorie werden die Orbitale, die in Richtung der Liganden zeigen, am stärksten beeinflusst. Je mehr bindende MOs nun mit Elektronen besetzt sind, desto stärker wird die Bindung zwischen Metall und Ligand und desto stabiler wird damit der Komplex. Mit 18 Valenzelektronen sind alle bindenden MOs besetzt und der Komplex hat die höchste Stabilität.
Die nebenstehende Grafik zeigt das MO-Diagramm des Ferrocens, welches mit 18 Elektronen besetzt ist. Die MO-Diagramme anderer Sandwichkomplexe sehen prinzipiell ähnlich aus, wenngleich die einzelnen Energieniveaus der Orbitale sich von Metall zu Metall unterscheiden. In Cobaltocen und Nickelocen sind die antibindenden e*1g-Orbitale mit einem bzw. zwei ungepaarten Elektronen besetzt, was zu einer Destabilisierung der M-Cp-Bindung und zu einer Aufweitung des M-C-Abstandes führt (Fe = 204 pm, Co = 211 pm, Ni = 218 pm).[45] Verändert sich die Geometrie des Komplexes oder die Ladung des Zentralatoms, kann sich dadurch ebenfalls die Reihenfolge der Molekülorbitale umkehren.[46]
Eigenschaften
BearbeitenNebengruppenmetallocene
Bearbeiten.JPG)
Von den Elementen der Gruppe 4 bis 12 (Nebengruppenelemente) existieren Dicyclopentadienyl-Komplexen in großer Anzahl. Einzig die Verbindungen der Gruppe 8 sind dabei 18-Elektronenkomplexe und daher elektronisch (besonders) stabil. Die Dicyclopentadienyl-Komplexe der anderen Gruppen erfüllen die 18-Elektronenregel nicht, was zur Folge hat, dass diese deutlich instabiler, bzw. reaktiver sind und nicht immer eine ideale Sandwichstruktur bilden. Die elektronenarmen Komplexe der Gruppen 4 bis 7 sind bestrebt, ihren Elektronenmangel durch weitere Liganden auszugleichen. Ohne weitere Reaktionspartner kann dies durch Dimerisierung oder Oligomerisierung geschehen. Die elektronenreichen Komplexe der 9. bis 12. Gruppe sind dagegen bestrebt, ihren Elektronenüberschuss zu reduzieren. Dies kann durch Oxidation oder durch eine Verringerung der Haptizität eines Liganden geschehen.
Von der 4. Periode existieren die Metallocene von Vanadium bis Nickel als isolierbare Verbindungen. Diese sind alle isomorph und haben einen Schmelzpunkt um 173 °C.[47] Von der 5. Periode sind nur die Metallocene von Ruthenium und Rhodium und von der 6. Periode nur das Osmocen isolierbare Verbindungen. Eine Übersicht über die bisher bekannten Metallocene bzw. Dicyclopentadienyl-Komplexe der Nebengruppenelemente gibt die folgende Tabelle:[48]
| Gruppe | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Valenz- elektronen[A 1] |
14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | ||
| 4. Periode | |||||||||||
| Kurzbezeichnung[A 2] Name Farbe Schmelzpunkt Siedepunkt M-C-Bindungsabstand[45] |
"TiCp2"[A 3] Titanocen grün 200 °C (Zers.) … … |
VCp2 Vanadocen purpur 167 °C … 227 pm |
CrCp2 Chromocen rot 173 °C … 215 pm |
MnCp2 Manganocen braun 173 °C 245 °C 211 pm |
FeCp2 Ferrocen orange 173 °C 249 °C 204 pm |
CoCp2 Cobaltocen purpur-schwarz 174 °C … 211 pm |
NiCp2 Nickelocen grün 173 °C … 218 pm |
[A 4] | "ZnCp2" Zinkocen farblos … … … | ||
| 5. Periode | |||||||||||
| Kurzbezeichnung Name Farbe Schmelzpunkt Siedepunkt M-C-Bindungsabstand |
"ZrCp2"[A 3] Zirconocen … … … … |
"NbCp2"[A 3] Niobocen gelb … … … |
MoCp2[A 5] Molybdocen schwarz … … … |
[A 6] | RuCp2 Ruthenocen hellgelb 195–200 °C … 221 pm[46] |
RhCp2 Rhodocen gelb 174 °C … … |
"CdCp2" Cadmocen … … … … | ||||
| 6. Periode | |||||||||||
| Kurzbezeichnung Name Farbe Schmelzpunkt Siedepunkt M-C-Bindungsabstand |
"TaCp2"[A 3] Tantalocen … … … … |
"WCp2"[A 3] Wolframocen gelb oder grün … … … |
ReCp2 Rhenocen … … … … |
OsCp2 Osmocen weiß … … 219 pm,[49] 222 pm[46] |
IrCp2 Iridocen … … … … |
"PtCp2"[A 3] Platinocen … … … … |
[A 4] | "HgCp2" Mercurocen … … … … | |||
Anmerkungen A:
- ↑ rechnerische Anzahl der Valenzelektronen in einem η5,η5-Cp2Metall-Komplex.
- ↑ Wenn die Kurzbezeichnung in Anführungszeichen gesetzt ist, handelt es sich nicht um ein klassisches Metallocen (z. B. liegt keine Sandwichstruktur vor).
- ↑ a b c d e f Nicht als (reiner) Sandwichkomplex existent.
- ↑ a b Kupfer, Gold: bisher nur berechnet.
- ↑ kann nur bei Matrixstabilisierung nachgewiesen werden.
- ↑ Technetium ist ein radioaktives Element.
- Strukturen ausgewählter Nebengruppenmetallocene
-
 Titanocen
Titanocen -
 Zirconocen
Zirconocen -
 Vanadocen
Vanadocen -
 Chromocen
Chromocen -
 Polymere Struktur von Manganocen
Polymere Struktur von Manganocen -
 Ferrocen
Ferrocen -
 Temperaturabhängiges Gleichgewicht zwischen monomerem und dimerem Rhodocen
Temperaturabhängiges Gleichgewicht zwischen monomerem und dimerem Rhodocen -
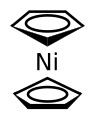 Nickelocen
Nickelocen







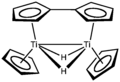
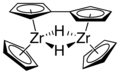
Gruppe 4: Titanocen existiert ebenso wie das homologe Zirconocen bei Raumtemperatur nur als Dimer, in dem zwei Cyclopentadienyl-Liganden als Fulvalen und die beiden Titanatome zusätzlich über zwei 2-Elektronen-3-Zentren-Wasserstoffbrücken miteinander verbunden sind. Jedes Titanatom besitzt dadurch 16 Valenzelektronen. Durch sterisch anspruchsvollere Liganden wie Pentamethylcyclopentadienyl (Cp*) kann der monomere 14-Elektronenkomplex TiCp*2 synthetisiert werden, welcher in gestaffelter Konformation vorliegt.[50][51] Um die Reaktionen des freien Titanocen zu untersuchen, kann dieses in-situ z. B. aus TiCp2Cl2 durch Umsetzung mit Magnesium, Lithium oder Natriumamalgan synthetisiert werden.[52]
Gruppe 5: Vanadocen ist ein purpurfarbener, kristalliner, paramagnetischer Feststoff, der als 15-Elektronenkomplex instabil ist. Niobocen existiert bei Raumtemperatur nicht als monomerer Sandwichkomplex, sondern dimerisiert als [NbH(C5H5)(C5H4)]2. In dem Dimer hat jedes Niobatom neben den zwei η5-Cp-Liganden eine zusätzliche η1Bindung zu einem Ringkohlenstoffatom des anderen Zentralatoms und einen weiteren Hydrid-Liganden. Zusammen mit der zusätzlich Niob-Niob-Metallbindung hat jedes Metallatom 18-Valenzelektronen.[50]
Gruppe 6: Chromocen ist ein roter kristalliner Feststoff, welcher an der Luft und gegenüber Wasser sehr reaktiv ist und sich unter Umständen bei Luftkontakt spontan entzünden kann.[48][35] Es wird, auf silikatischen Trägersubstanzen aufgebracht, als Katalysator bei der Polymerisation von Ethylen und anderen 1-Alkenen eingesetzt.[53][54] Wie Molybdocen kann auch Wolframocen nur als reaktive Zwischenstufe, z. B. photochemisch aus WCp2H2 oder thermisch aus WCp2(H)CH3 hergestellt werden.[55] Beide Komplexe sind als Monomere bei Raumtemperatur nicht stabil und dimerisieren unter Bildung von verschiedenen isomeren Zweikernkomplexen.[46]
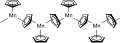
Gruppe 7: Manganocen ist ein brauner, oberhalb 158 °C rosafarbener Feststoff mit ungewöhnlichen magnetischen Eigenschaften. Aufgrund der günstigen high-spin-d5-Konfiguration (jedes d-Orbital ist mit einem Elektron besetzt), kann es nicht zu Mn+ reduziert werden, um die günstige 18-Elektronen-Konfiguration zu erhalten.[56] Im Festkörper liegt Manganocen polymer vor, jedes Mangan ist von drei Cyclopentadienyl-Liganden umgeben. Zwei der drei Liganden sind dabei mit jeweils zwei Manganzentren verbunden, während das dritte nur an ein Manganatom gebunden ist. Die verbrückenden Cyclopentadienyl-Liganden liegen dabei nicht symmetrisch zwischen den Manganatomen.[57] Rhenocen kann durch Photolyse von Re(C5H5)2H in einer Stickstoff- und Argonmatrix bei 12 K hergestellt, isoliert und untersucht werden. Unter diesen Bedingungen liegt der Komplex monomer vor und hat eine Sandwichstruktur.[58]
Gruppe 8: Ferrocen, das einzig luftstabile Metallocen, ist ein diamagnetischer 18-Valenzelektronenkomplex, in welchem das Eisenatom durch die Cp-Ringe sterisch gut abgeschirmt wird. Beim Erhitzen in einer evakuierten Glasampulle zersetzt es sich oberhalb 550 °C.[59] Aufgrund seiner außerordentlichen Stabilität und seines aromatischen Verhaltens (die Cyclopentadienylringe sind für elektrophile Substitutionsreaktionen zugänglich) war und ist Ferrocen Gegenstand zahlreicher Untersuchungen.[60] Osmocen ist im Vergleich zu Ferrocen und Ruthenocen weniger reaktiv gegenüber elektrophiler aromatischer Substitution, zeigt aber die größte Tendenz zur Bildung von Addukten mit Lewis-Säuren.[61]
Gruppe 9: Die 19-Elektronen-Komplexe Cobaltocen, Rhodocen und Iridocen werden leicht zum entsprechenden 18-Elektronen Metalloceniumkation oxidiert.[62] Die Oxidationsneigung ist dabei so groß, dass bereits organische Halogenide dazu ausreichen:

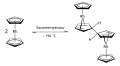
Cobaltocen dient entsprechend als 1-Elektronen-Reduktionsmittel und kann als Indikator für wasserfreie Redoxsysteme verwendet werden. Das Cobaltociuniumkation ist so stabil, dass der Di(methylcyclopentadienyl)-Komplex durch Salpetersäure nicht zerstört, sondern stattdessen die Methylgruppe am aromatischen Ring zur Carbonsäuregruppe oxidiert wird.[62] Rhodocen ist nur bei Temperaturen über 150 °C oder durch Kühlung auf die Temperatur von flüssigem Stickstoff (−196 °C) stabil. Bei Raumtemperatur (25 °C) wandelt sich Rhodocen in Acetonitril in weniger als 2 Sekunden durch Dimerisierung (Kombination) zu [Rh(C5H5)2]2, einem diamagnetischen 18-Valenzelektronen-Komplex um, in welchem zwei Rhodocen-Einheiten über Cyclopentadienylringe miteinander verbunden sind.[63][64][65][66] Dimeres Rhodocen [Rh(C5H5)2]2 ist ein gelber Feststoff.
Gruppe 10: Nickelocen ist ein dunkelgrüner, kristalliner Feststoff, der leidlich luftbeständig ist und sich leicht oxidieren lässt. Palladocen und Platinocen, die beiden anderen Komplexe der Gruppe 10, sind bislang nicht bekannt, lediglich die entsprechenden Dikationen konnten mit Hilfe von sperrigen Liganden, wie Cp* synthetisiert werden.[67][68]
Gruppe 11: Metallocene von Kupfer, Silber und Gold konnten noch nicht hergestellt werden; Cu(C5H5)2 und Au(C5H5)2 wurden bisher nur berechnet.[69]
Gruppe 12: Zinkocen wurde zuerst 1969 durch Ernst Otto Fischer beschrieben. Es polymerisiert in einer Kettenstruktur, in welcher abwechselnd Cyclopentadienylringe und Zinkatome miteinander verknüpft sind und jedes Zinkatom zusätzlich einen weiteren endständigen Cyclopentadienylring besitzt.[70][71] Cadmocen wurde zuerst 1969 von Jörg Lorberth beschrieben. Aufgrund seiner Unlöslichkeit in unpolaren Lösungsmitteln und der hohen Zersetzungstemperatur von > 250 °C wurde angenommen, dass es ebenfalls eine polymere Struktur besitzt.[72] Durch den Einsatz sperriger Substituenten wie iso-Propyl- oder tert-Butyl konnten von Dirk Bentz momomere Cadmocen-Derivate dargestellt werden, welche eine η1,η1 oder eine η1,η2-Koordinierung der Ringe aufweisen.[70]
Die relative Stabilität der Nebengruppenmetallocene kann bestimmt werden, indem die Redoxpotentiale der einfach geladenen Kationen gegenübergestellt werden. Die Daten der folgende Aufstellung werden gegenüber einer gesättigten Kalomel-Elektrode in Acetonitril bestimmt:
- [Fe(C5H5)2]+ / [Fe(C5H5)2] +0,38 V[73]
- [Co(C5H5)2]+ / [Co(C5H5)2] −0,94 V[63]
- [Rh(C5H5)2]+ / [Rh(C5H5)2] −1,41 V[63]
Diese Daten zeigen die Stabilität des neutralen Ferrocens sowie der Cobaltocenium- und Rhodocenium-Kationen. Rhodocen wirkt um etwa 500 mV stärker reduzierend als Cobaltocen, was auch bedeutet, dass es sich leichter oxidieren lässt und entsprechend weniger stabil ist.[63] Frühere polarografische Untersuchungen an Rhodoceniumperchlorat bei einem neutralen pH-Wert zeigten an einer tropfenden Quecksilber-Elektrode ein Signal bei −1,53 V (gegenüber einer gesättigten Kalomel-Elektrode), entsprechend der Bildung von Rhodocen in Lösung; trotzdem war es den Forscher nicht möglich, das neutrale Rhodocen zu isolieren. In der gleichen Untersuchung versuchten sie Iridocen unter oxidierenden Bedingungen aus Iridoceniumsalzen nachzuweisen, was aber auch bei erhöhten pH-Werten nicht gelang.[17] Diese Ergebnisse zeigen, dass das Rhodocen sehr instabil ist, aber weisen auch darauf hin, dass das Iridocen sogar noch instabiler ist.
Cyclopentadienyl-Komplexe der Seltenen Erden
BearbeitenDie Metalle der Gruppe 3 (Seltene Erden) bilden in der Regel keine klassischen Sandwichkomplexe. Die Komplexe mit der allgemeinen Formel MCp3 werden durch Umsetzung der entsprechenden Halogenide mit Cyclopentadienylnatrium erhalten. Geoffrey Wilkinson und J. M. Birmingham synthetisierten und beschrieben 1956 eine ganze Reihe von Cyclopentadienyl-Komplexen der Seltenen Erden:[24]
| M(C5H5)3 M = |
Farbe | Schmelzpunkt | Farbe (geschmolzen) |
|---|---|---|---|
| Scandium | strohfarben | 240 | rot |
| Yttrium | schwach gelb | 295 °C | grünlich gelb |
| Lanthan | farblos | 395 °C[B 1] | farblos |
| Cer | orange-gelb | 435 °C[B 1] | gelb |
| Praseodym | schwach grün | 415 °C[B 1] | grün |
| Neodym | rotblau | 380 °C | rotblau |
| Samarium | orange | 415 °C | orange |
| Gadolinium | gelb | 350 °C | gelb |
| Dysprosium | gelb | 302 °C | gelb |
| Erbium | pink | 285 °C | pink |
| Ytterbium | dunkelgrün | 273 °C | grün[B 2] |
Anmerkungen B:
Alle Cyclopentadienyl-Komplexe der Seltenen Erden vom Typ MCp3 haben ähnliche chemische Eigenschaften. Sie sind in Petrolether, Cyclohexan und Benzol nicht und in Pyridin, THF, Ethylenglycoldimethylether und 1,4-Dioxan schwach löslich. Mit Wasser zersetzen sie sich rasch zum Metallhydroxid und Cyclopentadien. Auch beim Luftkontakt zersetzen sie sich rasch. Mit Eisen(II)-chlorid reagieren sie in THF rasch und quantitativ unter Bildung von Ferrocen. Das chemische Verhalten, sowie physikalische Eigenschaften, wie die magnetische Suszeptibilität (welche nahe bei den korrespondierenden Ionen sind) implizieren einen ionischen Charakter der Komplexe.
Allerdings können auch MCp2-Komplexe der seltenen Erden synthetisiert werden. Von diesen sind die nach den Lanthanoiden benannten Lanthanocen(II)komplexe von Samarium, Europium und Ytterbium am längsten bekannt. Diese neigen dazu, mit Lösungsmitteln wie THF Donorkomplexe von Typ Cp2(THF)2 zu bilden. E.O. Fischer synthetisierte 1964 die Komplexe EuCp2 und YbCp2 im flüssigen Ammoniak und reinigte diese durch Sublimation.[74] Lösungsmittelfreies SmCp*2 wurde 1984 von William J. Evans hergestellt.[75] 1986 konnte William J. Evans mittels Röngtenstrukturanalysen zeigen, dass SmCp*2 und EuCp*2 eine gewinkelte Sandwichstruktur mit einem Cp-M-Cp-Winkel von 140° besitzen.[76]
Cyclopentadienyl-Komplexe der Actinoide
BearbeitenDie Metalle aus der Reihe der Actinoiden bilden ebenso wie die Seltenenerdmetalle keine klassischen Sandwichkomplexe. Die Cp3An Komplexe sowie ihre Tetrahydrofuran-Addukte (Cp3An·thf) wurden in den Jahren zwischen 1965 und 1974 durch Salzmetathese mit Cyclopentadienylnatrium beziehungsweise durch Transmetallierung mit BeCp2 oder MgCp2 erhalten.[77]
| M(C5H5)3 M = |
Farbe | Schmelzpunkt |
|---|---|---|
| Thorium[C 1] | grün | – |
| Uran | braun | > 200 °C |
| Neptunium | braun | – |
| Plutonium | grün | 180 °C (Zersetzung) |
| Americium | fleischfarben | 300 °C (Zersetzung) |
| Curium | farblos | – |
| Berkelium | bernsteinfarben | – |
| Californium | rot | – |
Anmerkung C:
- ↑ Verbindung nicht vollständig charakterisiert
Die angegebenen Verbindungen können ebenfalls durch chemische Reduktion der vierwertigen Halogenid-Komplexe Cp3AnX (X = Halogenid) dargestellt werden, beispielsweise mit Natrium-Amalgam:

Alkalimetallocene
Bearbeiten
Die erste Alkalicyclopentadienylverbindung, Kaliumcyclopentadienid, wurde bereits 1901 von Johannes Thiele hergestellt, dessen Struktur konnte aber erst 1997 durch R. E. Dinnebier und F. Olbrich aufgeklärt werden.[78] Im Kristall bilden Kalium- und Cp-Ionen eine lineare Kettenstruktur, in der die Cp-Ringe jeweils gegeneinander abgewinkelt und die Kaliumatome zentral über den Ringen platziert sind.[78] Vergleichbare Strukturen findet man in den Kristallen von RbCp und CsCp. Im Gegensatz dazu bilden LiCp und NaCp eine ideale lineare Kette mit einer parallelen Anordnung der Cp-Ringe.[79][78][80] Die gemessenen Bindungswinkel zu den Cp-Ringen liegen im LiCp und NaCp bei 180° und verringern sich bei den schwereren Homologen: Kalium 138°, Rubidium 132° und Cäsium 130°.[78][80]
Im Lithoceniumion nehmen die beiden Cp-Ringe eine gestaffelte Konformation, mit einem Li-Cp-Abstand von 201 pm, ein.[81][82][83] Aufgrund der Schrägbeziehung im Periodensystem sind Lithocen-Komplexe in Struktur und Reaktionen dem weiter unten beschriebenen Magnesocen ähnlich. Organolithiumkomplexe haben neben der ionischen Bindung einen deutlich kovalenten Anteil, welcher zu einer starken Verzerrung der Struktur des Carbanions führen kann. Dieser kovalente Anteil nimmt bei den schwereren Homologen stark ab und bereits beim Kalium findet sich eine fast ausschließlich ionische Bindung.[29]
In neuerer Zeit wurden erste Natrocenium-Komplexe beschrieben. Deren Isolierung gelingt durch den Einsatz von Kronenethern, welche mit Natriumionen komplexe Kationen bilden.[79] Sandwichkomplexe der schwereren Homologen Kalium, Rubidium und Cäsium sind bisher nicht bekannt.
Erdalkalimetallocene
BearbeitenVon allen Elemente der Gruppe 2 (Erdalkalimetalle) wurden Biscyclopentadienyl-Komplexe synthetisiert.[32][30] Beryllocen wurde erstmals 1959 durch Ernst Otto Fischer aus Berylliumchlorid und Alkalicyclopentadienyl synthetisiert.[84] Magnesocen wurde bereits 1954 unabhängig voneinander von Ernst Otto Fischer und Geoffrey Wilkinson synthetisiert.[85][86] Es kann durch Disproportionierung von CpMgBr bzw. durch Reaktion von metallischem Magnesium mit Cyclopentadien bei 500 °C gewonnen werden. Die Biscyclopentadienyl-Komplexe von Calcium, Strontium und Barium wurden 1961 ebenfalls von Ernst Otto Fischer erstmals beschrieben. Sie können durch Umsetzung von metallischem Calcium oder Strontium mit Cyclopentadien in THF oder DMF oder durch Reaktion der entsprechenden Hydride von Calcium, Strontium und Barium mit Cyclopentadien gewonnen werden:[30] Die Cokondensation von metallischen Barium mit Cyclopentadien bei −196 °C liefert eine nahezu quantitative Ausbeute.[87]

Von den Erdalkalimetallocenen hat nur Magnesocen die klassische Sandwichstruktur.[28][88] E. Weiß konnte zeigen, dass Magnesocen im Kristall eine gestaffelte Konformation mit einem Mg-C-Abstand von 230 pm hat; dagegen ist nach A. Haland in der Gasphase die Metall-Cp-Bindung aufgeweitet und das Molekül liegt in ekliptischer Konformation vor.[28][32] Ob die Bindung zwischen Metall und Cp-Ring eher kovalenter oder eher ionischer Art ist, ist noch nicht klar. Die dem Ferrocen analoge Sandwichstruktur lässt nicht zwingend auf eine kovalente Bindung schließen, sie könnte auch durch Van-der-Waals-Wechselwirkungen erklärt werden. Für den eher ionischen Charakter sprechen die elektrische Leitfähigkeit in flüssigem Ammoniak, die heftige Hydrolysereaktion sowie die 13C-NMR-Verschiebung von 108 ppm (zum Vergleich LiCp = 103,6 ppm; NaCp = 103,4 ppm, FeCp2 = 68,2 ppm).[88][28] Dagegen sprechen 25Mg-NMR-Daten eher für eine weitgehend kovalente Bindung.[28]

Beryllocen zeigt je nach Aggregatzustand unterschiedliche Molekülgeometrien. Im festen Zustand zeigt es eine slipped-Sandwich-Struktur, die Ringe sind gegeneinander versetzt – ein Ring ist η5 der zweite nur η1koordiniert (Be-Cp-Abstand: 181 pm).[89][90] In der Gasphase scheinen beide Ringe η5 koordiniert zu sein. Tatsächlich ist ein Ring deutlich weiter entfernt als der andere (190 und 147 pm) und die scheinbare η5-Koordination ist auf eine schnelle Fluktuation der Bindung zurückzuführen.[88] Der Grund für die η5,η1-Struktur ist, dass die Orbitale des Beryllocens nur mit max. 8 Valenzelektronen besetzt werden können.
Mit zunehmender Ordnungszahl steigt der ionische Charakter der Erdalkalimetallocene. Im Kristall hat Ca(C5H5)2 einen polymeren Aufbau, wobei jedes Zentralatom von vier Cp-Liganden umgeben ist.[91] Dabei haben die Cp-Ringe unterschiedliche Haptizität (η5-, η5-, η3-, η1-).[87] Wird als Ligand Pentamethylcyclopentadienyl (Cp*) eingesetzt, können in der Gasphase die Strukturen der isolierten Moleküle durch Elektronenbeugung ermittelt werden.[92] Überraschenderweise zeigt sich hierbei, dass die Moleküle eine abgewinkelte Struktur haben, in denen die Abwinkelung mit steigender Größe des Zentralatoms zunimmt: Mg = 180°, Ca = 154°, Sr = 149°, Ba = 148°.[82] Andere Untersuchungen von Richard Blom u. a. zeigten bei den Pentamethylcyclopentadienyl-Komplexen CaCp*2 und YbCp*2, dass die Ebenen um 20° gegeneinander geneigt sind.[93] Für die Abwinkelung wurden verschiedene Modelle herangezogen:[92][80]
- elektrostatisches Modell – die negativen Liganden stören die Kugelsymmetrie der Elektronenhülle am Zentralatom
- Van-der-Waals Wechselwirkung – Triebkraft für die Abwinklung ist der Gewinn an Van-der-Waals-Anziehung zwischen den Liganden
- (n-1)d-Orbital Beteiligung – Bildung von ds-Hybridorbitalen
- ab-initio MO Methoden
Werden sterisch anspruchsvollere Ligangen wie Pentaisopropylcyclopentadienyl (C5iPr5)verwendet, so wird die Abwinkelung wieder aufgehoben (Beispiel: Ba(C5iPr5)2: 180°).[87]
Magnesocen und die schwereren Homologe bilden mit sauerstoff-, stickstoff- und phosphorhaltigen Lewis-Basen entsprechende Addukte.[32] Magnesocen bildet mit Ammoniak oder Aminen 1:1- oder 1:2-Komplexe, welche sich im Falle von primären oder sekundären Aminen isolieren und kristallographisch untersuchen lassen.[94] In diesen Addukten verändert sich die Haptizität eines Cp-Ringes von η5 auf η2.[79][95] Mit 2 THF-Molekülen bildet sich dagegen ein η5,η1-Komplex.[95][32] Der entsprechende Komplex von Barocen mit 2 THF ist dann wiederum ein η5,η5-Komplex.[87]
Verwendung
BearbeitenAufgrund ihrer unterschiedlichen chemischen Eigenschaften und Reaktivität finden die Metallocene und Metallocenderivate in der Forschung und in der Praxis vielfältige Verwendung. So wird Magnesocen im Labor für den Transfer von Cyclopentadienyl-Liganden auf andere Metalle verwendet.[28]
Als Metallquelle
BearbeitenThermisch oder photochemisch instabile Metallocene finden als Quelle für hochreinen Metalldampf Verwendung. So wird Magnesocen zur Beschichtung von Nanopartikeln[96] und im ALE-Verfahren (atomic layer epitaxy), bei denen z. B. grüne oder blaue LEDs hergestellt werden, eingesetzt.[32] Auch wird es als Dotierungsmittel bei der Herstellung von p-dotierten Halbleitern im CVD-Verfahren (chemical vapour deposition) verwendet.[94][97] Barocen kann im ALD-Prozess (atomic layer deposition) zum Auftragen von BaTiO3-Dünnschichten für Widerstände und Kondensatoren eingesetzt werden.[87] Zur Verringerung des Verbrauchs kann Ferrocen Diesel oder Heizöl beigemischt werden, um eine bessere Sauerstoffbindung und somit eine effektivere und sauberere Verbrennung (Reduzierung von Rußpartikeln) zu erreichen.[98]
Als Polymerisationskatalysatoren
BearbeitenDie heute mit Abstand wichtigste Anwendung für Metallocene und deren Derivate ist ihr Einsatz als Polymerisationskatalysatoren zur Herstellung von Polyolefinen.[99]
Seit den 1950er Jahren werden Ziegler-Natta-Katalysatoren zur Polymerisation von Olefinen, wie Ethylen oder Propylen bei niedrigen Drücken und Temperaturen eingesetzt. Dabei wird in einem mehrstufigen Additions-Insertions-Mechanismus das Olefin zunächst an einen titanorganischen Komplex angelagert und dann in die Titan-Kohlenstoff-Bindung eingebaut. An die dabei freiwerdende Koordinationsstelle wird dann das nächste Olefin angelagert und die Kettenreaktion schreitet durch den Einbau des Olefins in die Titan-Kohlenstoffbindung weiter fort. Die klassischen Ziegler-Natta-Katalysatoren sind Mischkatalysatoren, welche aus einer metallorganischen Hauptgruppen-Verbindung der Gruppen I, II oder III (z. B. Triethylaluminium) und einer Übergangsmetallverbindung, hauptsächlich der Gruppen IV bis VI (z. B. Titantetrachlorid), bestehen. Sie haben jedoch den entscheidenden Nachteil, dass sie in der Regel als heterogene Katalysatoren auf einem Trägermaterial eingesetzt werden, da sie in organischen Lösungsmitteln nicht löslich sind. Dadurch spielen neben den eigentlichen Katalysatoreigenschaften auch die Eigenschaften des Trägermaterials, die Diffusionsgeschwindigkeit des Olefins und andere Adsorptionreaktionen eine Rolle. 1982 entdeckte Patricia Wilson, dass das lösliche LuCp*2CH3 als Polymerisationskatalysator für Ethylen und Propylen auch ohne einen Coinitiator fungiert.[100]

1980 beschrieben Hansjörg Sinn und Walter Kaminsky die katalytische Polymerisationsreaktionen von Mischungen der Metallocendihalogenide (Typ 1) mit Methylaluminoxan (MAO). Diese Kaminsky-Katalysatoren erlauben die Polymerisation von Ethylen, Propylen oder Olefin-Mischungen mit sehr hoher Produktivität und Selektivität.[101] Während Metallocene mit einem konventionellen Aluminiumalkyl-Cokatalysator nur eine geringe Aktivität zeigen, erhöht sich durch die Anwesenheit von Methylaluminoxan im Überschuss deren Reaktivität um den Faktor 10 000 und mehr, wodurch diese hundertfach aktiver sind als die traditionellen Ziegler-Natta-Katalysator. Auch sind Kaminsky-Katalysatoren in Kohlenwasserstoffen löslich und können daher, anders als Ziegler-Natta-Katalysatoren, direkt in Lösung eingesetzt werden.[102] Mit Kaminsky-Katalysatoren auf der Basis von Zirconocen können bis zu 100 Tonnen Ethylen pro Gramm Zirkonium polymerisiert werden, wobei die Insertionszeit in der Größenordnung von 10−5 s liegt.[102]
Fünf Jahre später beschrieben Walter Kaminsky und Hans-Herbert Brintzinger, dass bei der Verwendung von ansa-Metallocenen vom Typ 2 (C2-symmetrische ansa-Metallocene) Polypropylen mit streng isotaktischer Anordnung hergestellt werden kann.[103] Durch Vergrößerung der organischen Reste an den Cp-Ringen (wie in Typ 3) und/oder Variationen der Brückenatome lassen sich Aktivität und Selektivität gezielt beeinflussen sowie die die Molekulargewichtsverteilung der entstehenden Polymere in engen Grenzen optimieren.[99][102]
Magnesocen und die Biscyclopentadienyl-Verbindungen von Calcium und Strontium können als Polymerisationskatalysator z. B. für Methacrylsäuremethylester (MMA) eingesetzt werden.[94][104]
In der Tumortherapie
Bearbeiten

Viele Metallocen-Derivate der frühen Übergangsmetalle haben keine klassische Sandwichstruktur. Stattdessen ist die Cp-M-Cp-Achse oft auf rund 130° abgewinkelt, da sie bis zu drei weitere Liganden tragen. Dadurch ist deren Zentralatom für Reaktionspartner leichter zugänglich und reaktiver, was sich unter anderem auch in einer erhöhten Bioaktivität bemerkbar machen kann. So zeigen die Titanocen-, Molybdocen-, Niobocen, Vanadocen- und Rhenocen-Derivate, vom Typ MCp2Cl2 eine zytostatische Wirkung.[105] In Untersuchungen wurde nachgewiesen, dass insbesondere Derivate von Titanocendichlorid TiCp2Cl2 in der Tumortherapie gegenüber Cisplatin eine verbesserte Wirksamkeit bei deutlich geringerer Toxizität besitzen.[106][107][108] Zusätzlich zeigen diese Wirksamkeit auch bei Krebsarten, welche im Laufe der Therapie Resistenzen gegenüber Cisplatin entwickelt können, was eine weitere Therapie bei erneutem Auftauchen von Krebszellen erschwert.[109] Andere Untersuchungen zeigten, dass kationische Komplexe von Niobocendichlorid und Molybdocendichlorid im Vergleich zu Titanocendichlorid eine weiter verbesserte Wirksamkeit haben.[105] Bisher ist noch kein Metallocen zur Behandlung von Krebserkrankungen zugelassen. Vom onkologisch vielversprechendsten Metallocen, dem Titanocendichlorid, endeten die Klinischen Studien in der Phase II. Bisher wurde wegen der zu geringen Aktivität noch keine Phase-III-Studie durchgeführt.[110]
In der Sensortechnik
BearbeitenAufgrund der herausragenden strukturellen Stabilität des Ferrocens, sowohl in seiner neutralen Form als auch als Ferroceniumkation, eignet sich Ferrocen hervorragend sowohl für die Messung von Redoxpotentialen als auch als Partner in Redoxreaktionen. Fixiert man z. B. eine Kombination eines Ferrocenderivates mit dem Enzym Glucose-Oxidase auf einer Elektrodenoberfläche, so lässt sich bei Anwesenheit von Glucose in einer physiologischen Flüssigkeit beim Anlegen eines elektrischen Potentials ein Strom messen. Durch den Vergleich mit einer Eichkurve lässt sich daraus z. B. bei Diabetikern direkt der Glucosegehalt im Blut bestimmen. Bindet man an Ferrocen eine funktionelle Gruppe, welche Kationen komplexiert, so verändert sich in Gegenwart eines Kations das Redoxverhalten dieses Ferrocenderivates. Reagiert die funktionelle Gruppe (ausschließlich) mit einem bestimmten Kation, so erhält man einen selektiven Sensor für dieses Kation.[45] In neuerer Zeit finden Ferrocenderivate auch Anwendung in nichtlinearen Optiken.[45]
Literatur
Bearbeiten- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 101. Auflage. Walter de Gruyter, Berlin 1995, ISBN 3-11-012641-9, S. 1699–1703.
- Walter Kaminsky: Metallocenes. In: Ullmann’s Encyclopedia of Industrial Chemistry. 15. Juni 2006, doi:10.1002/14356007.b16_b36.pub2 (englisch).
- Anthony F. Hill: Organotransition Metal Chemistry. Royal Society of Chemistry, 2002, ISBN 978-0-85404-622-5, S. 161 f., doi:10.1039/9781847551597. (zur Erläuterung des Begriffes "ansa-Metallocene")
Weblinks
BearbeitenEinzelnachweise
Bearbeiten- ↑ Eintrag zu Cyclopentadienyl. In: Römpp Online. Georg Thieme Verlag, abgerufen am 8. Juni 2014.
- ↑ Press Release: The Nobel Prize in Chemistry 1973. The Royal Swedish Academy of Sciences, 1973, abgerufen am 28. Dezember 2011.
- ↑ a b T. J. Kealy, P. L. Pauson: A New Type of Organo-Iron Compound. In: Nature. Band 168, Nr. 4285, 1951, S. 1039–1040, doi:10.1038/1681039b0.
- ↑ a b c Peter L. Pauson: Ferrocene—how it all began. In: J. Organomet. Chem. Band 637–639, 2001, S. 3–6 (caltech.edu [PDF; 103 kB]).
- ↑ a b Samuel A. Miller, John A. Tebboth, John F. Tremaine: Dicyclopentadienyliron. In: J. Chem. Soc. 1952, S. 632–635, doi:10.1039/JR9520000632.
- ↑ a b c d F.Albert Cotton: Cyclopentadienyl–metal chemistry in the Wilkinson Group, Harvard, 1952–1955. In: J. Organomet. Chem. Band 637–639, Dezember 2001, S. 18–26, doi:10.1016/S0022-328X(01)01130-5.
- ↑ Geoffrey Wilkinson, M. Rosenblum, M. C. Whiting, R. B. Woodward: The Structure of Iron Bis-cyclopentadienyl. In: J. A. Chem. Soc. 1952, S. 2125–2126, doi:10.1021/ja01128a527.
- ↑ Pierre Laszlo, Roald Hoffmann: Ferrocen: objektive Geschichte oder eine Rashomon-Erzählung? In: Angewandte Chemie. Band 112, Nr. 1, 2000, S. 127–128, doi:10.1002/(SICI)1521-3757(20000103)112:1<127::AID-ANGE127>3.0.CO;2-2.
- ↑ P. F. Eiland, R. Pepinsky: X-ray Examination of Iron Biscyclopentadienyl. In: J. Am. Chem. Soc. Band 74, Nr. 19, 1952, S. 4971, doi:10.1021/ja01139a527.
- ↑ J. Dunitz, L. Orgel, A. Rich: The crystal structure of ferrocene. In: Acta Crystallographica. Band 9, Nr. 4, 1956, S. 373–375, doi:10.1107/S0365110X56001091.
- ↑ R. B. Woodward, M. Rosenblum, M. C. Whiting: A NEW AROMATIC SYSTEM. In: J. Am. Chem. Soc. Band 74, Nr. 13, 1952, S. 3458–3459, doi:10.1021/ja01133a543.
- ↑ G. Wilkinson: The Preparation and Some Properties of Ruthenocene and Ruthenicinium Salts. In: J. Am. Chem. Soc. Band 74, Nr. 23, 1952, S. 6146–6147, doi:10.1021/ja01143a538.
- ↑ G. Wilkinson, P. L. Puason, J. M. Birmingham, F. A. Cotton: Bis-cyclopentadienyl Derivates of some Transition Elements. In: J. Am. Chem. Soc. Band 75, Nr. 4, 1953, S. 1011–1012, doi:10.1021/ja01100a527.
- ↑ E. O. Fischer, R. Jira: Di-cyclopentadienyl-nickel. In: Zeitschrift für Naturforschung B. 8, 1953, S. 217–219 (PDF, freier Volltext).
- ↑ E. O. Fischer, R. Jira: Di-cyclopentadienyl-kobalt(II). In: Zeitschrift für Naturforschung B. 8, 1953, S. 327–328 (online).
- ↑ E. O. Fischer, W. Hafner: Di-cyclopentadienyl-chrom. In: Zeitschrift für Naturforschung B. 8, 1953, S. 444–445 (online).
- ↑ a b c F. A. Cotton, R. O. Whipple, G. Wilkinson: Bis-Cyclopentadienyl Compounds of Rhodium(III) and Iridium(III). In: J. Am. Chem. Soc. Band 75, Nr. 14, 1953, S. 3586–3587, doi:10.1021/ja01110a504.
- ↑ a b G. Wilkinson: Cyclopentadienyl Compounds of Chromium, Molybdenum and Tungsten. In: J. Am. Chem. Soc. Band 76, Nr. 1, 1954, S. 209–211, doi:10.1021/ja01630a053.
- ↑ J. M. Birmingham, A. K. Fischer, G. Wilkinson: The Reduction of Bis-cyclopentadienyl Compounds. In: Naturwissenschaften. Band 42, Nr. 4, 1955, S. 96, doi:10.1007/BF00617242.
- ↑ G. Wilkinson, J. M. Birmingham: Biscyclopentadienylrhenium hydride – a new type of hydride. In: J. Am. Chem. Soc. Band 77, Nr. 12, 1955, S. 3421–3422, doi:10.1021/ja01617a098.
- ↑ G. Wilkinson, J. M. Birmingham: Bis-cyclopentadienyl Compounds of Ti, Zr, V, Nb and Ta. In: J. Am. Chem. Soc. Band 76, Nr. 17, 1954, S. 4281–4284, doi:10.1021/ja01646a008.
- ↑ L. Friedman, A. P. Irsa, G. Wilkinson: Mass Spectra of Cyclopentadienyl Metal Compounds. Part I. Bis-cyclopentadienyl Compounds of V, Cr, Fe, Co, Ni, Re and Ru, and Manganese and Magnesium Cyclopentadienides. In: J. Am. Chem. Soc. Band 77, Nr. 14, 1955, S. 3689–3692, doi:10.1021/ja01619a004.
- ↑ G. Wilkinson, J. M. Birmingham: cyclopentadienyl Compounds of Sc, Y, La, Ce and some Lanthanide elements. In: J. Am. Chem. Soc. Band 76, Nr. 23, 1954, S. 6210, doi:10.1021/ja01652a114.
- ↑ a b G. Wilkinson, J. M. Birmingham: The Cyclopentadienides of Scandium, Yttrium and Some Rare Earth Elements. In: J. Am. Chem. Soc. Band 78, Nr. 1, 1956, S. 42–44, doi:10.1021/ja01582a009.
- ↑ a b J. M. Birmingham, D. Seyferth, G. Wilkinson: A New Preparation of Bis-cyclopentadienyl-Metal Compounds. In: J. Am. Chem. Soc. Band 76, Nr. 16, 1954, S. 4179, doi:10.1021/ja01645a038.
- ↑ J. Huheey, E. Keiter, R. Keiter: Anorganische Chemie. de Gruyter Verlag, 1993, ISBN 3-11-017903-2 (Seite 800 in der Google-Buchsuche).
- ↑ Didier Astruc: Organometallic Chemistry and Catalysis. Springer-Verlag, Berlin, Heidelberg 2007, ISBN 978-3-540-46128-9 (Seite 251–270 in der Google-Buchsuche).
- ↑ a b c d e f g Christoph Elschenbroich: Organometallchemie. B. G. Teubner Verlag, 2008, ISBN 978-3-8351-0167-8 (Seite 65–71 in der Google-Buchsuche).
- ↑ a b c d Erwin Riedel: Moderne Anorganische Chemie. de Gruyter, 2007, ISBN 978-3-11-019060-1 (Seite 622 in der Google-Buchsuche).
- ↑ a b c d Ernst Otto Fischer, Georg Stölzle: Erdalkali-di-cyclopentadienyle des Calciums, Strontiums und Bariums. In: Chem. Ber. Band 94, Nr. 8, 1961, S. 2187–2193, doi:10.1002/cber.19610940836.
- ↑ J. Grobe, B. H. Schneider, H. Zimmermann: Darstellung. In: Z. Anorg. Allg. Chem. Band 481, Nr. 10, 1981, S. 107–116, doi:10.1002/zaac.19814811012.
- ↑ a b c d e f Anja Jaenschke: Basenaddukte des Magnesocens, Dissertation 2006 (pdf, 6.8 MB).
- ↑ A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 101. Auflage. Walter de Gruyter, Berlin 1995, ISBN 3-11-012641-9, S. 1701–1702.
- ↑ Albrecht Salzer: Nomenklatur metallorganischer Verbindungen der Übergangsmetalle. In: Angewandte Chemie. Band 114, Nr. 11, 2000, S. 2043–2058, doi:10.1002/1521-3757(20020603)114:11<2043::AID-ANGE2043>3.0.CO;2-3.
- ↑ a b A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 101. Auflage. Walter de Gruyter, Berlin 1995, ISBN 3-11-012641-9, S. 1699–1700.
- ↑ Didier Astruc: Organometallic Chemistry and Catalysis. Springer-Verlag, Berlin, Heidelberg 2007, ISBN 978-3-540-46128-9 (Seite 251–270 in der Google-Buchsuche).
- ↑ a b c Didier Astruc: Organometallic Chemistry and Catalysis. Springer-Verlag, Berlin, Heidelberg 2007, ISBN 978-3-540-46128-9 (Seite 251–270 in der Google-Buchsuche).
- ↑ Ulrich T. Mueller-Westerhoff: [m.m]Metallocenophane: Synthese, Struktur, Eigenschaften. In: Angewandte Chemie. Band 98, Nr. 8, 1986, S. 700–716, doi:10.1002/ange.19860980805.
- ↑ A. Lüttringhaus, W. Kullick: Ansa-Ferrocene. In: Angewandte Chemie. Band 70, Nr. 14, 1958, S. 438, doi:10.1002/ange.19580701407.
- ↑ Irene Resa, Ernesto Carmona, Enrique Gutierrez-Puebla, Angeles Monge: Decamethyldizincocene, a Stable Compound of Zn(I) with a Zn-Zn Bond. In: Science. Band 305, Nr. 5687, 2004, S. 1136–1138, doi:10.1126/science.1101356.
- ↑ Josef T. Boronski, Agamemnon E. Crumpton, Lewis L. Wales, Simon Aldridge: Diberyllocene, a stable compound of Be(I) with a Be–Be bond. In: Science. Band 380, Nr. 6650, 2023, S. 1147–1149, doi:10.1126/science.adh4419.
- ↑ Inga-Alexandra Bischoff, Sergi Danés, Philipp Thoni, Bernd Morgenstern, Diego M. Andrada, Carsten Müller, Jessica Lambert, Elias C. J. Gießelmann, Michael Zimmer, André Schäfer: A lithium–aluminium heterobimetallic dimetallocene. In: Nature Chemistry. 2024, S. 1–8, doi:10.1038/s41557-024-01531-y.
- ↑ E. O. Fischer, W. Pfab: Zur Kristallstruktur der Di-Cyclopentadienyl-Verbindungen des zweiwertigen Eisens, Kobalts und Nickels. In: Z. Anorg. Allg. Chem. Band 274, Nr. 6, 1953, S. 316–322, doi:10.1002/zaac.19532740603.
- ↑ R. C. Mehrotra, A. Singh: Organometallic Chemistry: A Unified Approach. 2. Auflage. New Age International, New Delhi 2007, ISBN 978-81-224-1258-1 (Seite 261–267 in der Google-Buchsuche).
- ↑ a b c d Piero Zanelli: Inorganic Electrochemistry theory, practice and applications. Royal Society of Chemistry, 2003, ISBN 0-85404-661-5 (Seite 159–216 in der Google-Buchsuche).
- ↑ a b c d Christoph Elschenbroich: Organometallchemie. B. G. Teubner Verlag, 2008, ISBN 978-3-8351-0167-8 (Seite 457–458 in der Google-Buchsuche).
- ↑ J. Huheey, E. Keiter, R. Keiter: Anorganische Chemie. de Gruyter Verlag, 1993, ISBN 3-11-017903-2 (Seite 792 in der Google-Buchsuche).
- ↑ a b Christoph Elschenbroich: Organometallchemie. B. G. Teubner Verlag, 2008, ISBN 978-3-8351-0167-8 (Seite 452 in der Google-Buchsuche).
- ↑ J. C. A. Bobyens, D. C. Levendis, Michael I. Bruce und Michael L. Williams: Crystal structure of osmocene, Os(η-C5H5)2. In: Journal of Chemical Crystallography. Band 16, Nr. 4, 1986, S. 519–524, doi:10.1007/BF01161040.
- ↑ a b Christoph Elschenbroich: Organometallchemie. B. G. Teubner Verlag, 2008, ISBN 978-3-8351-0167-8 (Seite 459 in der Google-Buchsuche).
- ↑ Peter B. Hitchcock, Francesca M. Kerton, Gerard A. Lawless: The Elusive Titanocene. In: J. Am. Chem. Soc. Band 120, Nr. 39, 1998, S. 10264–10265, doi:10.1021/ja981934e.
- ↑ Thomas Janssen: Bis(<eta>1,<eta>5-pentafulven)titankomplexe: Reagenzien für selektive Reaktionen mit N–H-aciden Substraten und Katalysatoren in der intramolekularen Alkenhydroaminierung, Dissertation 2010 (PDF; 2,4 MB).
- ↑ Patent DE4306105A1: Modifizierte geträgerte Chromocen-Katalysatorsysteme.
- ↑ Manfred Dieter Lechner, Klaus Gehrke, Eckhard H. Nordmeier: Makromolekulare Chemie: ein Lehrbuch für Chemiker, Physiker, Materialwissenschaftler und Verfahrenstechniker. 4. Auflage. Birkhäuser, Basel 2010 (Seite 93 in der Google-Buchsuche).
- ↑ John F. Hartwig, Xiaoming He: Reaktivität von Wolframocen gegenüber B-B und B-H-Bindungen im Vergleich zu C-H-Bindungen. In: Angewandte Chemie. Band 108, Nr. 3, 1996, S. 352–354, doi:10.1002/ange.19961080321.
- ↑ Christoph Elschenbroich: Organometallchemie. B. G. Teubner Verlag, 2008, ISBN 978-3-8351-0167-8 (Seite 468 in der Google-Buchsuche).
- ↑ Erwin Riedel: Moderne Anorganische Chemie. de Gruyter, 2007, ISBN 978-3-11-019060-1 (Seite 707 in der Google-Buchsuche).
- ↑ Jeremy N. Hill, Robin N. Perutz, A. Denise Rooney: Laser-Induced Fluorescence of Rhenocene in Low-Temperature Matrixes: Selective Excitation and Emission. In: J. Phys. Chem. Band 99, Nr. 2, 1995, S. 531–537, doi:10.1021/j100002a014.
- ↑ Christian Müller: Grundlegende Untersuchungen zum CVD-Wachstum Fe-gefüllter Kohlenstoff-Nanoröhren ( vom 6. Juni 2014 im Internet Archive), Dissertation, 2007 (PDF; 15,4 MB).
- ↑ Didier Astruc: Organometallic Chemistry and Catalysis. Springer-Verlag, Berlin, Heidelberg 2007, ISBN 978-3-540-46128-9 (Seite 251–270 in der Google-Buchsuche).
- ↑ Sally A. Kur, Arnold L. Rheingold, Charles H. Winter: Synthesis, Characterization, and Halogenation of Decakis(acetoxymercurio)osmocene. Crystal and Molecular Structure of Decachloroosmocene. In: Inorg. Chem. Band 34, Nr. 1, 1995, S. 414–416, doi:10.1021/ic00105a067.
- ↑ a b A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 101. Auflage. Walter de Gruyter, Berlin 1995, ISBN 3-11-012641-9, S. 1622.
- ↑ a b c d N. El Murr, J. E: Sheats, W. E. Geiger, J. D. Holloway: Electrochemical Reduction Pathways of the Rhodocenium Ion. Dimerization and Reduction of Rhodocene. In: Inorg. Chem. Band 18, Nr. 6, 1979, S. 1443–1446, doi:10.1021/ic50196a007.
- ↑ E. O. Fischer, H. Wawersik: Über Aromatenkomplexe von Metallen. LXXXVIII. Über Monomeres und Dimeres Dicyclopentadienylrhodium und Dicyclopentadienyliridium und Über Ein Neues Verfahren Zur Darstellung Ungeladener Metall-Aromaten-Komplexe. In: J. Organomet. Chem. Band 5, Nr. 6, 1966, S. 559–567, doi:10.1016/S0022-328X(00)85160-8.
- ↑ H. J. Keller, H. Wawersik: Spektroskopische Untersuchungen an Komplexverbindungen. VI. EPR-spektren von (C5H5)2Rh und (C5H5)2Ir. In: J. Organomet. Chem. Band 8, Nr. 1, 1967, S. 185–188, doi:10.1016/S0022-328X(00)84718-X.
- ↑ B. De Bruin, D. G. H. Hetterscheid, A. J. J. Koekkoek, H. Grützmacher: The Organometallic Chemistry of Rh–, Ir–, Pd–, and Pt–Based Radicals: Higher Valent Species. In: Prog. Inorg. Chem. Band 55, 2007, ISBN 978-0-471-68242-4, S. 247–354, doi:10.1002/9780470144428.ch5 ([1]).
- ↑ Helmut Werner, Hans-Jürgen Kraus: Synthese und Strukturdynamik von Bis(cyclopentadienyl)phosphanpalladium-Komplexen. In: Angewandte Chemie. Band 91, Nr. 12, 1979, S. 1013–1014, doi:10.1002/ange.19790911221.
- ↑ Oleg V. Gusev, Tat'yana A. Peganova, Mikhail G. Peterleitner, Svetlana M. Peregudova, Larisa I. Denisovich, Nikolai A. Ustynyuk: Bis(η5-pentamethylcyclopentadienyl)-and(η5-cyclopentadienyl) (η5-pentamethylcyclopentadienyl)-platinium dications: Pt(IV) metallocenes. In: J. Organomet. Chem. Band 480, Nr. 1–2, 1994, S. c16–c17, doi:10.1016/0022-328X(94)87134-5.
- ↑ Diego Carrascal, Lucas Fernández-Seivane, and Jaime Ferrer: Oscillating spin-density pattern in gold metallocene and phthalocyanine molecules. In: Phy. Rev. B. Band 80, 2009, S. 184415, doi:10.1103/PhysRevB.80.184415.
- ↑ a b Dirk Bentz: Komplexe von Zink, Cadmiun, Lanthan, Cer und Samarium mit sperrigen Alkylcyclopentadienyl-Liganden. Dissertation 2005, urn:nbn:de:hbz:386-kluedo-19006
- ↑ Peter H.M. Budzelaar, Jaap Boersma, Gerrit J.M. van der Kerk, Anthony L. Spek, Albert J.M. Duisenberg: The structure of dicyclopentadienylzinc. In: J. Organomet. Chem. Band 281, Nr. 2–3, 1985, S. 123–130, doi:10.1016/0022-328X(85)87100-X.
- ↑ J. Lorberth: Organometallverbindungen der iib-elemente: Dicyclopentadienyl-zink, -cadmium und -quecksilber. In: J. Organomet. Chem. Band 19, Nr. 1, 1969, S. 189–190, doi:10.1016/S0022-328X(00)87767-0.
- ↑ V. V. Pavlishchuk, A. W. Addison: Conversion Constants for Redox Potentials Measured Versus Different Reference Electrodes in Acetonitrile Solutions at 25 °C. Band 298, Nr. 1. Inorg. Chim. Acta, 2000, S. 97–102, doi:10.1016/S0020-1693(99)00407-7.
- ↑ E. O. Fischer, H. Fischer: Über Dicyclopentadienyleuropium und Dicyclopentadienylytterbium und Tricyclopentadienyle des Terbiums, Holmiums, Thuliums und Lutetiums. In: J. Organomet. Chem. Band 3, Nr. 3, 1965, S. 181–187, doi:10.1016/S0022-328X(00)87500-2.
- ↑ William J. Evans, Laura A. Hughes, Timothy P. Hanusa: Synthesis and crystallographic characterization of an unsolvated, monomeric samarium bis(pentamethylcyclopentadienyl) organolanthanide complex, (C5Me5)2Sm. In: J. Am. Chem. Soc. Band 106, Nr. 15, 184, S. 4270–4272, doi:10.1021/ja00327a037.
- ↑ William J. Evans, Laura A. Hughes, Timothy P. Hanusa: Synthesis and x-ray crystal structure of bis(pentamethylcyclopentadienyl) complexes of samarium and europium: (C5Me5)2Sm and (C5Me5)2Eu. In: Organomet. Band 5, Nr. 7, 1986, S. 1285–1291, doi:10.1021/om00138a001.
- ↑ Carol J. Burns, Moris S. Eisen: Organoactinide Chemistry: Synthesis and Characterization. In: The Chemistry of the Actinide and Transactinide Elements. Band 5, 2008, Kap. 25, S. 2800–2805, doi:10.1007/1-4020-3598-5_25.
- ↑ a b c d Robert E. Dinnebier, Ulrich Behrens, Falk Olbrich: Solid State Structures of Cyclopentadienyllithium, -sodium, and -potassium. Determination by High-Resolution Powder Diffraction. In: Organomet. Band 16, Nr. 17, 1997, S. 3855–3858, doi:10.1021/om9700122.
- ↑ a b c Joachim Paap: Neuartige pi-Organyle der schweren Alkalimetalle und des Magnesiums ( vom 23. September 2015 im Internet Archive), Dissertation 2004 (pdf, 4.8 MB).
- ↑ a b c Robert E. Dinnebier: Kristallstrukturbestimmung molekularer Substanzen aus Röntgenbeugungsaufnahmen an Pulvern ( vom 14. Mai 2011 im Internet Archive), (pdf, 4,1 MB)
- ↑ Sjoerd Harder, Marc Heinrich Prosenc: The Simplest Metallocene Sandwich: the Lithocene Anion. In: Angew. Chem. Int. Ed. Band 33, Nr. 17, 1994, S. 1744–1746, doi:10.1002/anie.199417441.
- ↑ a b Dietmar Stalke: The Lithocene Anion and "Open" Calcocene–New Impulses in the Chemistry of Alkali and Alkaline Earth Metallocenes. In: Angew. Chem. Int. Ed. Band 33, Nr. 21, 1994, S. 2168–2171, doi:10.1002/anie.199421681., (pdf, 421 kB) ( vom 5. März 2016 im Internet Archive)
- ↑ Zvi Rappoport, Ilan Marek: The Chemistry of Organolithium Compounds. John Wiley & Sons Ltd., 2004, ISBN 0-470-84339-X (Seite 65–68 in der Google-Buchsuche).
- ↑ E. O. Fischer, H. P. Hofmann: Über Aromatenkomplexe von Metallen, XXV. Di-cyclopentadienyl-beryllium. In: Chem. Ber. Band 92, Nr. 2, 1959, S. 482–486, doi:10.1002/cber.19590920233.
- ↑ E. O. Fischer, W. Hafner: Cyclopentadienyl-vanadin-tetracarbonyl. In: Zeitschrift für Naturforschung B. 9, 1954, S. 503–504 (online).
- ↑ G. Wilkenson, F.A. Cotton, Chem. Ind. (London) 1954, S. 307–308.
- ↑ a b c d e Kai Fichtel: Metallocen-Verbindungen des Bariums ( vom 23. September 2015 im Internet Archive), Dissertation 2004 (pdf, 5,4 MB).
- ↑ a b c Erwin Riedel: Moderne Anorganische Chemie. de Gruyter, 2007, ISBN 978-3-11-019060-1 (Seite 623 in der Google-Buchsuche).
- ↑ Christoph Elschenbroich: Organometallchemie. B. G. Teubner Verlag, 2008, ISBN 978-3-8351-0167-8 (Seite 60 in der Google-Buchsuche).
- ↑ J. Huheey, E. Keiter, R. Keiter: Anorganische Chemie. de Gruyter Verlag, 1993, ISBN 3-11-017903-2 (Seite 799 in der Google-Buchsuche).
- ↑ J. Huheey, E. Keiter, R. Keiter: Anorganische Chemie. de Gruyter Verlag, 1993, ISBN 3-11-017903-2 (Seite 794 in der Google-Buchsuche).
- ↑ a b Christoph Elschenbroich: Organometallchemie. B. G. Teubner Verlag, 2008, ISBN 978-3-8351-0167-8 (Seite 70 in der Google-Buchsuche).
- ↑ R. A. Andersen, R. Blom, J. M. Boncella, C. J. Burns, H. V. Volden: The Thermal Average Molecular Structures of Bis(pentamethylcyclopentadienyl)magnesium(II), -calcium(II) and -ytterbium(II) in the Gas Phase. In: Acta Chem. Scand., Ser. A. Band 41, 1987, S. 24–35, doi:10.3891/acta.chem.scand.41a-0024.
- ↑ a b c Aibing Xia, John E. Knox, Mary Jane Heeg, H. Bernhard Schlegel, and Charles H. Winter: Synthesis, Structure, and Properties of Magnesocene Amine Adducts. Structural Distortions Arising from N-H…C5H5 - Hydrogen Bonding and Molecular Orbital Calculations Thereof. In: Organomet. Band 22, 2003, S. 4060–4069. (pdf, 260 kB)
- ↑ a b Zvi Rappoport, Ilan Marek: The Chemistry of Organomagnesium Compounds, Band 2. John Wiley & Sons Ltd., 2008, ISBN 978-0-470-05719-3 (Seite 124–129 in der Google-Buchsuche).
- ↑ Patent EP1073514: Method for the production of coated particles. Veröffentlicht am 7. Februar 2001, Erfinder: Dieter Vollath, Vinga Szabo, Bernd Seith.
- ↑ Patent US7449404: Method for improving Mg doping during group-III nitride MOCVD. Veröffentlicht am 11. November 2008, Erfinder: J. Randall Creighton, Goerge T. Wang.
- ↑ Patent EP0325769: Use of an unleaded liquid fuel containing ferrocen for operating a spark ignition engine. Veröffentlicht am 2. August 1989, Erfinder: Dieter Höhr, August-Wilhelm Preuss, Kurt-Peter Schug, Helmut Riegel.
- ↑ a b Michael Aulbach, Frank Küber: Metallocene – maßgeschneiderte Werkzeuge zur Herstellung von Polyolefinen. In: Chemie in unserer Zeit. Band 28, Nr. 4, 1997, S. 197–208, doi:10.1002/ciuz.19940280410.
- ↑ Didier Astruc: Organometallic Chemistry and Catalysis. Springer-Verlag, Berlin, Heidelberg 2007, ISBN 978-3-540-46128-9 (Seite 367–373 in der Google-Buchsuche).
- ↑ Hansjörg Sinn, Walter Kaminsky, Hans-Jürgen Vollmer, Rüdiger Woldt: „Lebende Polymere“ bei Ziegler-Katalysatoren extremer Produktivität. In: Angewandte Chemie. Band 92, Nr. 5, 1980, S. 396–402, doi:10.1002/ange.19800920517.
- ↑ a b c Walter Kaminsky: Metallocenes. In: Ullmann's Encyclopedia of Industrial Chemistry. Wiley, 15. Juni 2006, S. 685–691, doi:10.1002/14356007.b16_b36.pub2 (englisch).
- ↑ Walter Kaminsky, Klaus Külper, Hans H. Brintzinger, Ferdinand R. W. P. Wild: Polymerisation von Propen und Buten mit einem chiralen Zirconocen und Methylaluminoxan als Cokatalysator. In: Angewandte Chemie. Band 97, Nr. 6, 1985, S. 507–508, doi:10.1002/ange.19850970617.
- ↑ Alexandra Steffens: Erdalkalimetallkomplexe zur Polymerisation polarer Monomere, Dissertation, 2005 (PDF; 3,5 MB).
- ↑ a b P. Köpf-Maier und T. Klapötke: Antitumor activity of ionic niobocene and molybdenocene complexes in high oxidation states. In: Journal of Cancer Research and Clinical Oncology. Band 118, Nr. 3, 1992, S. 216–221, doi:10.1007/BF01410137.
- ↑ P. Köpf-Maier: Histologic and ultrastructural alterations of a xenografted human colon adenocarcinoma after treatment with titanocene dichloride. In: Journal of Cancer Research and Clinical Oncology. Band 114, Nr. 4, 1988, S. 250–258, doi:10.1007/BF00405830.
- ↑ P. Köpf-Maier: Tumor inhibition by titanocene complexes: influence on xenografted human adenocarcinomas of the gastrointestinal tract. In: Cancer Chemotherapy and Pharmacology. Band 23, Nr. 4, 1989, S. 225–230, doi:10.1007/BF00451646.
- ↑ P. Manohari Abeysinghe and Margaret M. Harding: Antitumour bis(cyclopentadienyl) metal complexes: titanocene and molybdocene dichloride and derivatives. In: Dalton Transactions. Band 32, 2007, S. 3474–3482, doi:10.1039/B707440A.
- ↑ Alexander Gmeiner: Beiträge zur Chemie von Übergangsmetallkomplexen mit Alkenylcarbenen, Phosphoryliden und bioaktiven Hydroxyverbindungen als Liganden, Dissertation 2010 (pdf, 7,1 MB).
- ↑ L. Wagner: Synthetische metallorganische Verbindungen als Apoptoseinduktoren. Dissertation, Medizinischen Fakultät Charité - Universitätsmedizin Berlin, 2012, S. 6.