Lysergsäureamide
Lysergsäureamide (meist kurz: Lysergamide) sind eine chemische Stoffgruppe, welche die Amide der Lysergsäure und ihre Derivate umfasst. Ihre Stammverbindung ist das Mutterkornalkaloid Ergin. Viele dieser Verbindungen weisen pharmakologische Eigenschaften auf, die sie als Arzneistoff oder auch Droge interessant machen. Lysergamidhaltige Arzneimittel werden vor allem in der Frauenheilkunde und Geburtshilfe, bei Migräne und Cluster-Kopfschmerz sowie bei Morbus Parkinson eingesetzt. Mit Blick auf den bekanntesten Vertreter der Gruppe – das hochpotente Psychedelikum Lysergsäurediethylamid (LSD) – spricht man im Falle synthetischer Lysergamide auch von LSD-Analoga. Dabei umfassen strukturelle LSD-Analoga alle Substanzen, deren chemische Struktur jener von LSD stark ähnelt. Ist die Ähnlichkeit so beschaffen, dass die Substanz im lebenden Organismus auch eine ähnliche Wirkung hervorruft wie LSD, hat man es überdies mit einem funktionellen LSD-Analogon zu tun. Diese Wirkung ist auch speziesabhängig und nicht unbedingt von Versuchstieren auf den Menschen übertragbar. Teilweise handelt es sich bei den funktionellen Analoga um Prodrugs, die nach Einnahme vom Körper in LSD umgewandelt werden. In manchen Fällen besitzen die Analoga eine eigenständige pharmakologische Aktivität. Neben wissenschaftlichem Interesse und industriellem Wirkstoffdesign besteht eine Motivation für die Entwicklung neuer LSD-Analoga darin, bestehende staatliche Regulierungen psychotroper Substanzen zu umgehen.
Übersicht über die Stoffgruppe
BearbeitenGrundstruktur
BearbeitenDie Struktur aller Lysergamide leitet sich von der Stammverbindung Ergin ab. Damit handelt es sich um Ergolene mit Doppelbindung zwischen den C-Atomen 9 und 10 (Δ9,10-Ergolene), die in Position 8 eine Carbonsäureamidgruppe aufweisen. Sie können daher als polycyclische Amide beschrieben werden, in die sowohl die Phenethylamin- als auch die Tryptaminstruktur eingebettet sind.[1]

|
Grundstruktur mit Nummerierung der Gerüstatome anhand der Ergolin-Struktur. Der Tryptamin-artige Teil der Struktur ist blau hervorgehoben, die Amidgruppe rot. Der Phenethylamin-artige Teil der Struktur führt vom Ring A direkt über die C-Atome 10 und 5 zur Aminogruppe (N-6). Das Molekül besitzt zwei Stereozentren (C-5 und C-8). Die dargestellte Stereochemie entspricht jener der natürlich vorkommenden D-(+)-Lysergsäure. Die Substituenten an Stickstoffatomen sind entsprechend der Anlage zum deutschen NpSG (siehe unten) nummeriert. Bei LSD ist R1=H, R2=Me und R3=R4=Et. |
Natürlich vorkommende Lysergamide
Bearbeiten
Bei einem großen Teil der Mutterkornalkaloide – nicht jedoch den Clavinen und Lysergsäuren – handelt es sich um Lysergamide, welche in einfache Lysergamide und Ergopeptide unterteilt werden. Einfache Lysergamide tragen an der Amidgruppe keinen weiteren oder nur eine kleine Hydroxyalkylgruppe als Substituenten. Hierzu gehören die Stammverbindung Ergin sowie Lysergsäurehydroxyethylamid und Ergometrin. Bei Ergopeptiden sind über die Amidgruppe Tripeptide an das Ergin-Grundgerüst gebunden. Durch die vielfältigen Kombinationsmöglichkeiten von zwei der drei Aminosäuren (die dritte ist fast immer Prolin)[2] zusätzlich zu den verschiedenen Substitutionsmustern am Ergin-Grundgerüst gibt es eine große Anzahl bekannter Ergopeptide.[3] Man unterscheidet zunächst Ergopeptine und Ergopeptame: erstere enthalten einen Oxazolidring, in welchem die Stellung 2' oxidiert ist, in letzteren bilden die letzten zwei Aminosäuren einen Dizyklus.[4] Bei Peptinen werden anhand der ersten Aminosäure weitere Untergruppen definiert, namentlich die Ergotamin- (Alanin), die Ergotoxin- (Valin) und die Ergoxin-Gruppe (α-Aminobuttersäure).[5]
Synthetische Lysergamide
BearbeitenKünstlich hergestellte Derivate leiten sich von natürlich vorkommenden Lysergamiden meist durch veränderte Substituenten an den drei Stickstoffatomen der Stammverbindung ab.[6] Das bekannteste Beispiel ist das N,N-Diethylderivat von Lysergamid, LSD. Nur wenige Lysergamide mit Substituentenmodifikationen an anderen Stellen des Ergolen-Gerüsts sind von Bedeutung, zu nennen sind hier lediglich die 2-Brom-Derivate des Ergopeptins Ergocryptin, Bromocriptin, bzw. von LSD, BOL-148. Welche synthetischen Lysergamide als „LSD-Analoga“ bezeichnet werden, wird auch in der wissenschaftlichen Literatur uneinheitlich gehandhabt. Während sich Strukturanaloga schon durch bloße Ähnlichkeit der chemischen Struktur als solche qualifizieren, weisen funktionelle Analoga darüber hinaus wie LSD eine halluzinogene Wirkung auf. Dessen ungeachtet werden sowohl manche nicht-halluzinogene LSD-Derivate wie BOL-148 als auch Lysergamide, die keine Diethylamide sind, als „LSD-Analoga“[7] oder „LSD-Kongenere“[8] bezeichnet, wo dies zweckmäßig erscheint.
Forschungsgeschichte
Bearbeiten
Spätestens seit der zweiten Hälfte des 15. Jahrhunderts wurde Mutterkorn in der Frauenheilkunde und Geburtshilfe eingesetzt, insbesondere als Mittel bei Wehenschwäche sowie zur Verminderung von Blutungen während und nach der Geburt. Im 17. Jahrhundert wurde es als Ursache des „Antoniusfeuers“ (siehe unten) erkannt.[9] Auf der Suche nach den Wirkstoffen im Mutterkorn gelang es dem französischen Pharmazeuten Charles Joseph Tanret anno 1875[10][11] als erstem,[12] mit „Ergotinin“ einen homogenen Bestandteil des komplexen Alkaloidgemischs zu isolieren, der aber wenig bis keine pharmakologische Wirksamkeit besaß und sich später als Gemisch dreier Lysergamide herausstellte.[13] Auch das 1906 gleichzeitig von George Barger und Francis Howard Carr[14][15] in England sowie F. Kraft[16] in der Schweiz abgetrennte, pharmakologisch hochwirksame „Ergotoxin“ bzw. „Hydroergotinin“ wurde 1943[17] von den Schweizer Chemikern Arthur Stoll und Albert Hofmann als Lysergamidgemisch erkannt und in seine Einzelbestandteile aufgeteilt.[18] Stoll war es auch, der bereits 1918[19][20] mit Ergotamin tatsächlich das erste Mutterkornalkaloid in Reinsubstanz gewonnen hatte.[21] Ergometrin wurde in der ersten Hälfte der 1930er Jahre in mehreren Laboratorien zugleich entdeckt.[12] Beim Versuch seiner Partialsynthese aus Isolysergsäure[22] synthetisierte Hofmann 1938 im Labor von Stoll bei Sandoz erstmals – als 25. Lysergamidderivat aus seiner Synthesereihe – „LSD-25“, das sich im Weiteren aber als weder zur medikamentösen Wehenförderung noch als Analeptikum geeignet erwies, weswegen es vorerst nicht weiter verfolgt wurde.[23] Patentiert und als Arzneimittel auf den Markt gebracht wurde von Sandoz in der Zwischenzeit das Ergometrin-Derivat Methylergometrin.[24] Als Hofmann im Jahre 1943 noch einmal LSD synthetisierte und ihn dabei trotz vorsichtigen Arbeitens rauschhafte Symptome befielen, führte er einen Selbstversuch mit einer – nach damaligem Wissensstand – sehr geringen Menge seines Produkts durch, das sich dabei als psychotrope Substanz nie dagewesener Potenz herausstelle.[23]
Seit der Entdeckung der psychotropen Wirkung von LSD wurden zahlreiche Derivate der Lysergsäure synthetisiert und charakterisiert, um seinen Wirkmechanismus zu erforschen. Nachdem es in den Jahren 1948 bis 1953 gelungen war, den Neurotransmitter Serotonin zu isolieren, zu identifizieren und darzustellen,[25] beobachtete die Gruppe um John H. Gaddum, dass LSD in glatter Muskulatur als Antimetabolit von Serotonin wirkt.[26] Vor diesem Hintergrund veranlasste die psychotomimetische Wirkung von LSD D. W. Woolley, E. Shaw und andere dazu, dem Zusammenhang zwischen der Rolle von Serotonin im Gehirn und der Entstehung von Psychosen wie Schizophrenie nachzugehen.[27][28] Frühzeitig fiel auf, dass das LSD-Derivat 2-Brom-LSD (BOL-148) Serotonin antagonisiert und keinerlei psychedelische Effekte hervorruft.[29] Daraufhin spielten LSD und seine Derivate eine wichtige Rolle in jener Phase der Psychopharmakologie, die von Albert Sjoerdsma und Michael G. Palfreyman als „Renaissance“ (1953–1970) der Serotoninforschung charakterisiert wurde.[25] Im Laufe der 1960er kam die Forschung zum Einsatz von LSD im Rahmen „psycholytischer“[30] und „psychedelischer Therapie“[31] weitgehend zum Erliegen, was oft mit der zur gleichen Zeit einsetzenden Prohibitionspolitik begründet wird. Der Medizinhistoriker Matthew Oram weist aber für die USA darauf hin, dass LSD-Forschung niemals verboten, teilweise sogar weiterhin gefördert wurde, der Niedergang vielmehr bereits früher einsetzte und auf komplexe Veränderungen der rechtlichen Rahmenbedingungen für Pharmaforschung allgemein – u. a. infolge des Contergan-Skandals – zurückzuführen ist.[32] Weiterhin wurden und werden Lysergamide einschließlich LSD zur biochemischen Grundlagenforschung verwandt und Derivate ohne ausgeprägte psychotrope Wirkung als Medikamente eingesetzt und erforscht.[23] In jüngerer Vergangenheit hat auch das akademische Interesse an psychotropen Lysergamiden zu möglichem therapeutischen Einsatz und für die neuropsychologische Forschung wieder zugenommen.[33]
Medizinische Verwendung
Bearbeiten
Bei der Mutterkorn-Behandlung von Frauen mit Menstruationsbeschwerden sowie während und nach schwierigen Geburten diente wegen seiner gebärmutterstimulierenden Wirkung insbesondere das Mutterkornalkaloid Ergometrin als Wirkstoff, welches aufgrund dessen von der Weltgesundheitsorganisation auch als unentbehrliches Arzneimittel geführt wird.[34] Ähnliche Wirkung zeigt sein synthetischer Abkömmling Methylergometrin. Ihre einstige Bedeutung auf diesem Gebiet haben die Lysergamide aber mittlerweile verloren.[35] Während Ergometrin selbst nicht mehr im Handel ist,[36] wurden vom Methylergometrin-Präparat Methergin in Deutschland im Jahr 2011 noch 0,8 Millionen Definierte Tagesdosen (DDD) verschrieben, bei rückläufiger Verschreibungshäufigkeit.[37] Die Zweckentfremdung von Ergotpräparaten zur Selbstabtreibung birgt die Gefahr schwerwiegender Nebenwirkungen für die ungewollt Schwangere in Form von vaskulärem Ergotismus (siehe unten).[38]
Mehrere Lysergamide werden als Mittel gegen Migräne eingesetzt oder erprobt. Das in der Migräneprävention effektive Methysergid verschwand infolge des Auftretens seltener, aber schwerer Nebenwirkungen (v. a. Retroperitonealfibrose) seit Mitte der 1960er wieder vom Markt.[39][40] Ergotamin und sein Dihydroderivat werden darüber hinaus zur präventiven Behandlung von Cluster-Kopfschmerz eingesetzt, 2-Brom-LSD wird als möglicher Arzneistoff hierfür untersucht.[41] Vom Dihydroergotamin-Präparat Ergotam-CT wurden 2013 in der BRD 1,7 Millionen DDD verschrieben, bei rückläufiger Verschreibungshäufigkeit.[42]
Als drittes bedeutsames Gebiet kommen Lysergamidderivate in der Therapie der Parkinson-Krankheit zum Einsatz. Bromocriptin-Präparate wurden in der BRD 2019 im Umfang von insgesamt 1,1 Millionen DDD verschrieben. Auf den 9,10-hydrierten Wirkstoff Cabergolin entfielen weitere 0,93 Millionen DDD.[43]
| Wirkstoff | Gesamtverordnungen (in Tausend) |
Anteil Generika an Verordnungen |
Gesamtnettokosten (in Tausend Euro) |
Jahr der Erhebung |
|---|---|---|---|---|
| Bromocriptin | 46,1 | 90,0 % | 1673,5 | 2017[44] |
| Cabergolin * | 59,6 | 30,9 % | 5614,5 | 2017[44] |
| Dihydroergotamin * | 37,1 | 100,0 % | 630,2 | 2013[45] |
| Methylergometrin | 63,7 | 0,0 % | 778,2 | 2011[46] |
Pharmakologie
BearbeitenAllgemeine Pharmakodynamik
BearbeitenDas pharmakologische Profil von Lysergamiden ist komplex, da sie mit einer Vielzahl von G-Protein-gekoppelten Rezeptoren wechselwirken, die biogene Amine als physiologische Liganden haben.[47][48]

Die psychedelische Wirkung von LSD und Wirkungsanaloga wird vermittelt über die Aktivierung kortikaler Serotonin-Rezeptoren des Typs 2A (5-HTR2A),[49] die überwiegend intrazellulär vorkommen.[50] Eine Schlüsselrolle kommt der indirekten Ausschüttung von Glutamat, insbesondere im präfrontalen Cortex zu,[51] wobei die vermittelnden Signaltransduktionskaskaden noch nicht in allen Einzelheiten aufgeklärt sind. fMRT-Studien zeigen, dass unter LSD-Einwirkung einerseits die Integrität funktionaler Netzwerke im Gehirn vermindert ist und die Signalvarianz innerhalb solcher Netzwerke abnimmt, andererseits verstärkt Hirnareale miteinander kommunizieren, die sonst untereinander nur wenig funktional verbunden sind. So begünstigt die intensivere funktionale Vernetzung anderer Hirnareale mit dem primären visuellen Cortex das Entstehen der charakteristischen Halluzination. Der Zusammenhang zwischen zunehmender Konnektivität im thalamokortikalen System und sensorischer Filterung im Thalamus muss noch genauer erforscht werden.[49]
Für die psychedelische Wirkung ist die Reizübertragung über den Gαq-PLC-Pfad entscheidend sowie das Überschreiten einer Efficacy-Schwelle, die im Bereich von 70 bis 74 % liegt. Zu dieser Schlussfolgerung kam eine Studie aus dem Jahr 2023. Sie maß die G-Protein-Dissoziation per BRET und referenzierte die Emax-Werte auf Serotonin. LSD als starkes Halluzinogen wies eine Emax von 99 % auf. Partialagonisten mit Werten kleiner als 70 %, wie 2-Brom-LSD (53 %) und Lisurid (63 %), zeigen keine Anzeichen psychedelischer Wirkung, und zwar ungeachtet der Aktivität anderer Transduktoren (Reizübertrager). Die Studie widerspricht der Spekulation, β-Arrestin2 könne als Transduktor für die Halluzinogenese eine prominente Rolle spielen.[52][53] β-Arrestin2 dürfte vielmehr für die Tachyphylaxie von Bedeutung sein, möglicherweise auch für eine antipsychotische Wirkung.[52] Die Frage, warum manche hocheffiziente 5-HTR2A-Agonisten nicht oder nur schwach psychedelisch wirken, wird mit dem Grad der Zellmembranpermeabilität erklärt. So wirken Agonisten, die aufgrund ihrer ausgeprägten Lipophilie zur passiven Diffusion imstande sind, psychedelisch, während höher polare Stoffe wie Serotonin, die die Membran nicht durchqueren, keinen solchen Effekt haben. Im Englischen wird diese Form der funktionellen Selektivität location bias genannt.[50]
Zahlreichen 5-HTR2A-Agonisten wohnt das Potential rasch depressionslösender Wirkung inne. Ergebnisse präklinischer Studien deuten darauf hin, dass sich der antidepressive Wirkanteil vom psychedelischen entkoppeln lässt. Dies wurde etwa am Beispiel des 5-HTR2A-Partialagonisten 2-Brom-LSD gezeigt, welches die psychedelische Wirkung von LSD und DOI hemmt und antidepressive Eigenschaften hat.[54] Der antidepressive Effekt wird zum Teil der Aktivierung der 5-HT2A-Rezeptoren zugeschrieben,[55] während andererseits erkannt wurde, dass LSD wie auch Psilocin unmittelbar an den Tyrosinkinaserezeptor Typ B (TrKB; als Dimer) hochaffin binden.[56] TrKB gilt als zentraler Vermittler antidepressiver Wirkung.[57] Diese Wirkstoffe erhöhen die Verfügbarkeit von TrkB an der Zelloberfläche und verstärken als Positivmodulatoren dessen Signalfunktion. Synergistisch bewirkt LSD die Hochregulierung und damit die erhöhte Verfügbarkeit des Neurotrophins BDNF, dem endogenen Liganden des TrKB.[56]

Sowohl LSD als auch Ergotamin wirken am 5-HT2B-Rezeptor als arrestinbevorzugende Agonisten,[58][33][59] aktivieren also vorrangig den β-Arrestin-Signalweg gegenüber der Phosphoinositidkaskade.[Anm. 1] Am 5-HT1B-Rezeptor tritt dagegen keine vergleichbare funktionelle Selektivität auf.[58] Die migräneprophylaktische Wirkung von Methysergid kommt über 5-HT2B-Antagonismus zustande, die Wirkung gegen akute Migräne von Ergotamin und Dihydroergotamin dagegen über 5-HT1B/1D-Partialagonismus. Agonistische bzw. partialagonistische Wirkung vieler Lysergamidderivate wie Bromocriptin an Dopamin-Rezeptoren vor allem der D2-ähnlichen Gruppe ist die Grundlage ihres Einsatzes in der Therapie der Parkinson-Krankheit, der ein Dopaminmangel zugrunde liegt.[60] In Kombinationstherapie mit der Dopamin-Vorstufe Levodopa kann es einen unerwünschten Nebeneffekt des langfristigen Levodopa-Einsatzes lindern, nämlich die davon induzierte Steigerung des Dopamin-Abbaus im Striatum, welche mit der Zeit die Dopamin-ersetzende Wirkung von Levodopa reduziert.[61] Auch gibt es Hinweise darauf, dass Dopamin-abhängige Vorgänge für einen zeitverzögert eintretenden Teil der LSD-Wirkung verantwortlich sind.[62] Darüber hinaus wechselwirken Lysergamide mehr oder weniger stark mit verschiedenen Adrenozeptoren. Historisch geht deren Unterteilung in α- und β-Typen darauf zurück, dass erstere von Mutterkornalkaloiden beeinflusst werden und letztere nicht. Aktivierung von α-Adrenozeptoren der glatten Muskulatur – wie im Uterus oder in Blutgefäßen – führt zu deren Kontraktion. Die absoluten und relativen Affinitäten zum α1- und α2-Rezeptorsubtyp stellen sich für verschiedene Lysergamide unterschiedlich dar.[63]
Die geringe Bindungsselektivität ist typisch für Mutterkornalkaloide, die deswegen im Vergleich zu den ebenfalls auf Serotonin-Rezeptoren abzielenden Triptanen als Dirty Drugs gelten.[64] Die relativen Affinitäten zu den verschiedenen Rezeptoren können bei Analoga in unterschiedlichem Ausmaß variieren; so hat man zwischen den Affinitäten verschiedener Lysergamide zu Dopamin-Rezeptoren der D1-ähnlichen Gruppe einerseits und zu Serotonin-Rezeptoren der 5-HT2-Familie andererseits keine nennenswerte Kovarianz gefunden. Dies eröffnet die Möglichkeit, durch geeignete Substitutionen die Selektivität synthetischer Lysergamide zu beeinflussen.[65]
Im Falle vieler funktioneller LSD-Analoga ist nicht gesichert, ob ihre pharmakologische Wirkung auf eine eigenständige Rezeptoraffinität zurückzuführen ist, oder ob es sich um eine Prodrug handelt, aus der erst in vivo LSD oder ein anderes wirksames Lysergamid freigesetzt wird. Beide möglichen Wirkmechanismen schließen sich auch nicht gegenseitig aus, allerdings ist es unwahrscheinlich, dass eine Prodrug, die bereits teilweise zum hochpotenten LSD metabolisiert wurde, mit einer etwaigen eigenen Aktivität nennenswert zur psychotropen Wirkung beiträgt, wofür sie mit dem hochaffinen LSD um die vorhandenen 5-HT2A-Rezeptoren konkurrieren müsste,[66] welches sich überdies durch eine lange Verweildauer ebenda auszeichnet.[33] Dazu passt, dass die auf dem Drogenmarkt erhältlichen LSD-Analoga N-Allyl-nor-LSD (AL-LAD), 1-Propionyl-LSD (1P-LSD) und N-Ethyl-nor-LSD (ETH-LAD) im Rahmen einer Konsumentenselbstauskunftstudie tendenziell als etwas schwächere Version von LSD bewertet wurden.[67]
Struktur-Wirkungsbeziehung
Bearbeiten-
 Dopamin
Dopamin -
 Noradrenalin
Noradrenalin -
 Adrenalin
Adrenalin -
 Serotonin
Serotonin




Grundlage der Struktur-Wirkungsbeziehung der Lysergamide ist, dass sie in ihrer chemischen Struktur mehreren Neurotransmittern ähneln. In ihrem Ergolen-Gerüst vereinen sie wesentliche Strukturmerkmale der Katecholamine (Dopamin, Noradrenalin, Adrenalin), die ebenfalls Phenylethylamine sind, sowie von Serotonin (5-Hydroxytryptophan), das gleichsam zu den Tryptaminen gehört.[1] Aminerge Rezeptoren weisen eine schmale, mit hydrophoben Seitenketten ausgekleidete orthosterische Bindungstasche für ihre physiologischen Transmitter auf, in die sich auch das Ergolen-Gerüst einfügt.[33]
Bedeutung der Amidsubstituenten
BearbeitenBesonders intensiv erforscht wird der Mechanismus der Wechselwirkung von Lysergamiden mit Serotonin-Rezeptoren. Dabei zeigt sich, dass für diese Liganden neben der orthosterischen Bindungstasche selbst auch der vorgelagerte Bereich von großer Bedeutung ist. In dieser erweiterten Bindungstasche (extended binding pocket, EBP) hält sich bei Bildung des Ligand-Rezeptor-Komplexes die C-8-ständige Amidgruppe mit ihren Substituenten auf.[59] Auch für die Spezifität der Bindung an Dopamin-Rezeptoren hat sich die Wechselwirkung mit der erweiterten Bindungstasche als bedeutsam herausgestellt.[68][69]
_model_for_LSD.svg)

Von entscheidender Bedeutung für die psychotrope Potenz des LSD ist seine namensgebende N,N-Diethylamidgruppe. Bereits geringe Modifikationen an dieser Stelle – wie beim Isomer mit je einer Methyl- und n-Propylgruppe statt zweier Ethylgruppen – führen zu deutlich verringerter Wirkung.[71] In einer homologen Reihe von N-Alkyl-N-iso-propyllysergamiden zeigt sich ein besonders starker Rückgang der Wirksamkeit im Tierversuch beim Übergang von der N-Ethyl-N-iso-propyl- zur N,N-Di-iso-propylverbindung.[72] Untersuchungen mit Lysergamiden mit amidisch gebundenen 2,4-Dimethylazetidinen, welche als erstarrte Modelle der N,N-Diethylamidgruppe dienen, geben Hinweise auf die Bedeutung der Konformation dieser Gruppe: Lediglich das Derivat mit (S,S)-(+)-2,4-Dimethylazetidin rief im Tierversuch eine ähnlich starke Wirkung hervor wie LSD, die beiden anderen Stereoisomere mit (R,R)-(−)- und cis-2,4-Dimethylazetidin dagegen nicht. In vitro durchgeführte Ligandenbindungstests zeigten hier auch hohe Affinitäten zum 5-HT2A-, 5-HT2B- und 5-HT2C-Rezeptor sowie die höchste intrinsische Aktivität bezüglich der Umsetzung von Phosphoinositid als Schritt der Signaltransduktion.[70] Auf Grundlage kristallographischer, kinetischer und molekulardynamischer Analysen wird dies damit erklärt, dass die Ethylgruppen im rezeptorgebundenen Zustand auf diese Weise trans zueinander stehen und nur diese Konformation weitere Wechselwirkungen des tiefer in den Rezeptor hineinragenden Ergolen-Gerüsts mit den Aminosäuren des Proteins ermöglicht, die den Liganden festhalten. Ist der Ligand an der Einnahme dieser fixierenden Konformation sterisch gehindert oder fehlen ihm die Substituenten, diffundiert er bereits nach kurzer Zeit wieder von der Bindungsstelle des Rezeptors fort. Die Kinetik der Rezeptor-Ligand-Bindung wirkt sich auf deren weitere Folgen nicht nur quantitativ, sondern auch qualitativ aus: je kürzer die Verweildauer an der Bindungsstelle von 5-HT2A- und 5-HT2B-Rezeptoren, desto geringer wird spezifisch der β-Arrestin2-Signalweg aktiviert.[33] In diesem Zusammenhang erweist sich auch eine der extrazellulären Schleifen (EL 2) des 5HT2A/2B-Rezeptors als bedeutsam, da sie eine Art beweglichen „Deckel“ bildet, der sowohl den Ein- als auch den Austritt von Liganden verlangsamt. Wechselwirkungen seiner Seitenketten mit den Substituenten in der erweiterten Bindungstasche können die Verweildauer weiter erhöhen.[33][73]
In vergleichenden kryoelektronenmikroskopischen Untersuchungen an Ligand-Rezeptor-Komplexen hat sich gezeigt, dass die Wechselwirkung zwischen C-8-ständigen Substituenten und erweiterter Bindungstasche auch wesentlich dafür ist, dass Bromocriptin viel stärker an Dopamin-Rezeptoren vom Subtyp D2 bindet als an solche vom Subtyp D1, da dessen große Tripeptidgruppe – unter anderem aufgrund der sterisch anspruchsvollen Leucin-Seitenkette – im Falle des D1-Subtyps in Konflikt mit der Seitenkette eines evolutionär nicht-konservierten Lysin sowie mit der Position einer transmembranären α-Helix (TM6) steht. Im Falle des D2-Subtyps ist erstere nicht vorhanden und letztere anders positioniert, sodass auch der große Ergopeptidsubstituent Platz findet.[69]
Bedeutung des Ergolengerüsts
Bearbeiten| R2 | ED50 / μmol kg−1 |
Ki / nM [3H]-5-HT |
Ki / nM [3H]-ketanserin |
Ki / nM [125I]-(R)-DOI |
|---|---|---|---|---|
| Allyl | 0,013 | 15,5 | 8,1 | 3,4 |
| Et | 0,020 | 3,8 | 5,1 | 5,1 |
| nPr | 0,037 | 4,9 | 5,6 | |
| Me | 0,046 | 5,9 | 5,2 | 5,1 |
| CH2cPr | 0,067 | 10,9 | 7,7 | |
| iPr | 0,10 | 21,4 | 14,1 | |
| nBu | 0,36 | 45,7 | 5,2 | |
| CH2C2H | 0,62 | 91,2 | 100,0 | |
| CH2CH2Ph | k. S. | |||
| H | k. S. | 30,2 | 158,0 | |
| ED50-Werte aus DD-Versuchen mit Ratten („k. S.“: keine Substitution, d. h. Ratten erkennen es nicht als LSD-analog), Hemmungskonstanten Ki aus Versuchen zur Verdrängung radioaktiv markierter Liganden von 5-HT2-Rezeptoren. Kleinere Zahlenwerte entsprechen höheren Potenzen (ED50) bzw. Rezeptoraffinitäten (Ki). Bei LSD selbst ist R2=Me. | ||||
Wichtig für die Verankerung sowohl im Serotonin-[33] als auch im Dopamin-Rezeptor[68] ist eine Ionenbindung zwischen der basischen – und damit unter physiologischen Bedingungen protonierten – Trialkylamingruppe in Position 6 und der Carboxylatgruppe einer konservierten Aspartat-Seitenkette. Wird die Methylgruppe am N-6 durch andere organische Gruppen ersetzt, lässt sich in manchen Fällen im Rahmen von Drug-Discrimination-Versuchen[Anm. 2] eine niedrigere mittlere effektive Dosis (ED50) bestimmen, ab der Versuchstiere, die zuvor gelernt hatten, nach intraperitonealer Gabe von LSD einerseits und isotoner Salzlösung andererseits unterschiedliche Schalter zu betätigen, LSD-entsprechendes Verhalten zeigen (siehe Tabelle). Vor allem mit Allyl- (AL-LAD), aber auch Ethyl- (ETH-LAD) und n-Propylgruppen (PRO-LAD) am N-6 ergaben sich niedrigere ED50-Werte als bei LSD, mit iso-Propyl-, n-Butyl- und Propargylgruppe dagegen höhere.[75] Ohne Methylgruppe (Nor-LSD) erkannten die Versuchstiere die Substanz nicht mehr als LSD-Analogon; gleiches gilt bei ihrer Ersetzung durch die 2-Phenylethylgruppe, was im Falle von Opiumalkaloiden dagegen oft zu hoher Aktivität führt.[76]
Die Wechselwirkung zwischen dem Indolring und dem Rezeptor scheint weniger festgelegt zu sein. Mit LSD tritt im 5-HT2B-Modell eine Wasserstoffbrückenbindung des (nicht basischen) Indol-NH zum Rückgrat-O eines Glycin auf, mit Ergotamin dagegen zur OH-Gruppe einer Threonin-Seitenkette.[33] Von verschiedenen Ergolinen und Tryptaminen ist bekannt, dass Alkylsubstituenten am Indol-N die Affinität zum 5-HT2A-Rezeptor des Menschen verringern, nicht jedoch zu jenem der Ratte. Der Unterschied ist so stark, dass sich dabei das Verhältnis der 5-HT2A-Rezeptoraffinität zwischen den Spezies umkehrt: Während die N-1-unsubstituierten Verbindungen eine höhere Affinität zum menschlichen Rezeptor haben, haben die N-1-substituierten eine höhere zum Rezeptor der Ratte.[77] Acylsubstituenten am Indol-N verringern die Affinität von LSD-Derivaten zu den meisten Monoamin-Rezeptoren einschließlich des 5-HT2A-Rezeptors um ein bis zwei Zehnerpotenzen. Die immer noch beträchtliche Potenz der N-1-acetylierten (ALD-52), -propionylierten (1P-LSD) und -butyrylierten (1B-LSD) Verbindungen ist auf effektive Deacylierung in vivo zurückzuführen.[66]
Allgemein zu einem Verlust der halluzinogenen Wirkung führen Inversionen eines der beiden oder beider Stereozentren (C-5, C-8), Reduktion der Δ9,10-Doppelbindung oder Substituenten am C-2, insbesondere Halogene.[6]
Pharmakokinetik
BearbeitenIm Körper beginnt die Biotransformation der nur schlecht wasserlöslichen Lysergamide in der Leber mit Funktionalisierungsreaktionen (Phase-I-Reaktionen). Von zentraler Bedeutung sind hier Oxidationsreaktionen, die von Enzymen des Cytochrom-P450-Systems katalysiert werden, vor allem von CYP3A4: CYP3A4 entfernt Alkyl- und Allylgruppen vom Amin-N (N-6), deethyliert Amidgruppen zum N-Monoethylamid und hydroxyliert das Ergolen-Gerüst. Außer bei N-1-acylierten LSD-Derivaten und ETH-LAD werden solche Hydroxylierungen auch von CYP1A2 katalysiert. Andere Isoenzyme der CYP-Superfamilie spielen nur eine kleine Rolle: CYP2C19 katalysiert von den untersuchten Muttersubstanzen nur die Demethylierung von LSD, CYP2C9 die Amid-Deethylierung nur bei LSD und ECPLA, CYP2D6 die Hydroxylierung von ALD-52, 1B-LSD und ETH-LAD.[78] Von CYP2D6 gibt es Polymorphismen, die einen nennenswerten Einfluss auf die pharmakokinetischen Parameter von LSD haben.[79] Acylgruppen am Indol-N (N-1) werden hydrolytisch durch Amidasen abgespalten.[78] Vor allem hierdurch kann LSD aus N-1-acylierten LSD-Derivaten freigesetzt werden.[66] Auch Alkylgruppen am Indol-N können in vivo rasch entfernt werden; so wird etwa Methysergid im Zuge eines First-Pass-Effekts zu einem großen Teil zu Methylergometrin N-1-demethyliert.[80]
In Phase II der Biotransformation werden die so funktionalisierten Metabolite glucuronidiert,[78] wodurch ihre Hydrophilie stark zunimmt und sie über die Niere mit dem Harn ausgeschieden werden können.
Toxikologie
Bearbeiten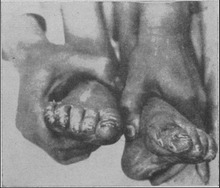
Mutterkornvergiftungen sind die wohl ältesten bekannten Mykotoxikosen.[81][9] Je nach Herkunft kann sich die Alkaloidzusammensetzung im Sklerotium unterscheiden und unterschiedliche Symptombilder hervorrufen. Man unterscheidet nach den Symptomen vor allem konvulsiven Ergotismus mit tonisch-klonischen Krampfanfällen und Halluzinationen sowie gangränösen (vaskulären) Ergotismus mit Gewebsnekrosen infolge von Blutunterversorgung aufgrund extremer Blutgefäßverengung.[82][83] Hinzu können Verdauungsbeschwerden (Enteroergotismus) und Überwärmung (hyperthermer Ergotismus) treten.[84] Werden gleichzeitig Medikamente eingenommen, die die CYP3A4 hemmen, können auch therapeutisch übliche Dosen von Lysergamid-Wirkstoffen giftig wirken, da ihr Abbau dadurch behindert wird. Mehrere Fälle sind bekannt, bei denen nach gleichzeitiger Einnahme eines Präparats mit 1 mg Ergotamin und eines CYP3A4-Hemmers gangränöser Ergotismus auftrat.[85] Im Vergiftungsfalle werden zur Therapie standardmäßig Vasodilatantien intraarteriell injiziert, um der gefährlichen Vasokonstriktion zu begegnen.[86] Auch längerdauernde Einnahme von Ergotaminen scheint das Ischämie-Risiko zu erhöhen, doch ist die Studienlage hierzu noch unzureichend.[87]
Für Ergopeptide – nicht jedoch für einfache Lysergamide wie Ergometrin – wurde ferner Zytotoxizität in menschlichen Zellkulturen demonstriert.[88]
Während von LSD selbst bekannt ist, dass es sich bei üblichen wirksamen Dosen (50–200 μg) um eine physiologisch äußerst sichere Substanz handelt,[89] gibt es zur Toxizität LSD-analoger neuer psychoaktiver Substanzen kein gesichertes Wissen,[78] sondern lediglich die Selbstauskunft von Konsumenten, Analoga als insgesamt etwas weniger wirksam und subjektiv noch weniger gefährlich als LSD wahrzunehmen.[67] Hinweise darauf, dass aktuell erhältliche Analoga eine signifikant höhere Toxizität hätten als LSD selbst, liegen bisher nicht vor.[90]
Chemie
BearbeitenBiosynthese
BearbeitenIhren Anfang nimmt die Biosynthese der Lysergamide in Mutterkornpilzen bei der Prenylierung der Aminosäure Tryptophan mit Dimethylallylpyrophosphat (DMAPP), katalysiert von der Prenyltransferase 4-Dimethylallyltryptophan-Synthase (DMATS). Nach Methyltransferase-katalysierter Methylierung der Aminogruppe mit S-Adenosylmethionin (SAM) wird über mehrere – ebenfalls enzymatische – Zwischenschritte zunächst Ring C und anschließend Ring D aufgebaut, wobei nach und nach verschiedene Clavine entstehen. Das Δ8,9-Ergolen Agroclavin wird in zwei weiteren Schritten enzymatisch zur Paspalsäure oxidiert, welche – entweder spontan oder mit Unterstützung seitens einer Isomerase – zur Lysergsäure tautomerisiert.[91]
.svg)
Wie im Weiteren die Lysergamide aus der D-Lysergsäure entstehen, ist ungewöhnlich: Eine nichtribosomale Peptidsynthetase (NRPS), die über zwei funktionell unterschiedliche Untereinheiten (D-Lysergylpeptidsynthetase 1 und 2) verfügt, aktiviert zunächst mit ihrer einen Untereinheit (LPS2) die Carboxylgruppe durch Bildung eines Thioesters. Nach Transfer an die andere Untereinheit (LPS1) katalysiert diese drei aufeinanderfolgende Kondensationsreaktionen mit Aminosäuren, wobei jedes Zwischenprodukt wieder als reaktiver Thioester an das Enzym gebunden bleibt. Nach Übertragung von Prolin als dritter Aminosäure kommt es zur Cyclokondensation mit der zuvor übertragenen Aminosäure und das Lactam – ein Ergopeptam – wird freigesetzt.[91] Dabei handelt es sich um das erste Beispiel eines NRPS-Multienzymkomplexes mit verschiedenen Untereinheiten, das in Pilzen gefunden wurde.[92] Die Weiterreaktion zu Ergopeptinen erfordert einen enzymatisch katalysierten Zwischenschritt, in dem die erste Aminosäure an ihrer α-Position oxidiert wird; das Zwischenprodukt cyclisiert spontan zum Oxazolidinon – zum Ergopeptin. Alternativ kann die aktivierte Lysergsäure mithilfe eines weiteren Enzyms (LPS3) mit der Aminosäure Alanin kondensieren und anschließend zu Ergometrin reduziert werden.[91]

Herstellung
BearbeitenBereitstellung des Grundgerüsts
BearbeitenDer übliche Weg zur Herstellung von Lysergamiden sind Partialsynthesen ausgehend von Lysergsäure, die in industriellem Maßstab (Weltjahresproduktion 1999 auf 10–15 t geschätzt)[93] durch Isomerisierung von Paspalsäure oder Hydrolyse anderer Mutterkornalkaloide aus Bioreaktoren mit saprophytisch kultivierten Mutterkornpilzen bereitgestellt wird.[94][95] Die natürlich vorkommenden Ergopeptine werden direkt durch Fermentation gewonnen, wobei deren geringe Wasserlöslichkeit bei dem dafür nötigen pH-Wert niedrige Konzentrationen in der Fermentationsbrühe bedingt.[96] Auch laborchemische Zugänge zum Aufbau des Ergolen-Gerüsts sind möglich: Der Forschungsgruppe um Robert B. Woodward gelang 1956 die erste, fünfzehnstufige Totalsynthese der Lysergsäure,[97] seitdem wurden zahlreiche weitere – auch enantioselektive – Zugänge entwickelt.[98] Diese Ansätze sind jedoch viel aufwändiger als die Isolierung aus natürlichem Material und anschließende Modifizierung.[99]
Die häufigsten Modifikationen in Lysergamiden betreffen die Substituenten an den Stickstoffatomen (Indol-N, Amin-N und Amid-N) sowie an Position 2 des Indolrings. Das Einführen von Substituenten an anderen Positionen – ohne die Δ9,10-Ergolen-Grundstruktur zu verändern – bereitet dagegen meist große präparative Schwierigkeiten.[6][100]
Synthese amidsubstituierter Lysergamide
BearbeitenDer erste synthetische Zugang zu Lysergamiden war die Spaltung natürlicher Ergopeptide mit Hydrazin, wodurch die Carbonsäurehydrazide der Diastereomere Lysergsäure und Isolysergsäure entstehen, deren Enantiomere durch Kristallisation mit Di-(p-toluyl)-D-weinsäure aufgetrennt werden können. Die Isolysergsäurehydrazide lagern sich in gesättigter Ethanol-Lösung nach Zugabe von Kalilauge basenkatalysiert zu den entsprechenden Lysergsäurehydraziden um.[101] Aus den Carbonsäurehydraziden können durch Umsetzung mit in situ gebildeter salpetriger Säure die Carbonsäureazide gewonnen werden. Die sehr reaktionsfähigen Lysergsäureazide bilden mit Aminen dann die entsprechenden Lysergsäureamide. Auch LSD wurde auf diese Weise erstmals synthetisiert.[22]

Diese Methode ist mit verschiedenen Nachteilen behaftet: Die Reaktionsbedingungen bewirken eine teilweise Racemisierung und Isomerisierung, was weitere Trennungs-, Umlagerungs- und Aufreinigungsschritte erfordert, die hochreaktiven Azide müssen in großen Mengen Lösungsmittel verdünnt werden, und der Acylierungsschritt nimmt mehrere Stunden in Anspruch. Dennoch waren lange Zeit keine besseren Zugänge verfügbar, da der klassische Weg der Amidsynthese – die Bildung der entsprechenden Carbonsäureester oder -chloride und die anschließende Umsetzung mit Aminen – bei Lysergsäure nicht funktioniert.[103] Im Laufe der Zeit wurden jedoch verschiedene Alternativrouten entwickelt, die die direkte Darstellung zahlreicher Lysergamide aus Lysergsäure ermöglichen, etwa über gemischte Säureanhydride mit Trifluoressigsäure oder Schwefelsäure,[103] mithilfe von Phosphorylchlorid,[102] oder mit PyBOP als Kupplungsreagenz.[70]
Synthese N-1-substituierter Lysergamide
BearbeitenModifikationen am Indol-N (Position 1) sind durch Alkylierungen, Acylierungen und Mannich-artige Reaktionen leicht zugänglich.[104] Alkylierungen werden mit primären Alkylhalogeniden und Kaliumamid in flüssigem Ammoniak durchgeführt.[105] Acylierungen sowie Sulfamoylierungen lassen sich mithilfe von Phasentransferkatalyse durch Umsetzung mit den entsprechenden Carbon- bzw. Sulfonsäurehalogeniden vornehmen.[106] Die Acetylierung mit Essigsäureanhydrid gelingt dagegen nicht, da es unter milden Bedingungen nicht mit Lysergsäure-Derivaten reagiert, unter energischeren Bedingungen aber der Ring D aufgespalten wird. Eine Alternative ist die Umsetzung mit Keten in Benzol mit katalytischen Mengen Triethylamin. Mannich-artige Kondensation von Dialkylaminen und Carbonylverbindungen ist mit Indolen möglich, wenn sie – wie die Derivate der Lysergsäure – an Position 3 Substituenten tragen. Hierdurch werden die N-1-Dialkylaminoderivate der Lysergamide zugänglich.[107] Diverse weitere Modifikationen sind möglich.[108]
Synthese C-2-substituierter Lysergamide
BearbeitenIn Position 2 ist die pharmazeutisch bedeutendste Modifikation das Einführen eines Brom-Atoms wie in Bromocriptin und 2-Brom-LSD (BOL-148). Direkte Bromierung mit elementarem Brom führt jedoch zur Überbromierung. Ein einzelnes Br-Atom kann mithilfe von N-Bromsuccinimid eingeführt werden, analog Iod mit N-Iodsuccinimid, Chlor mit N,2,6-Trichlor-4-nitro-acetanilid.[109] Speziell für die Synthese von Bromocriptin aus Ergocryptin wurden zahlreiche weitere Verfahren entwickelt.[110]
Synthese N-6-substituierter Lysergamide
Bearbeiten
Die Darstellung von am Amin-N (Position 6) anders substituierten Lysergamiden aus Lysergsäure erfordert zu Beginn deren N-Demethylierung. Da es sich beim Ausgangsstoff um ein tertiäres Amin handelt, kann es im Rahmen einer Von-Braun-Reaktion mit Bromcyan in Tetrachlormethan (unter Freisetzung von Methylbromid)[111] zur entsprechenden N-Cyanoverbindung umgesetzt werden, welche sich anschließend mit Zink in verdünnter Essigsäure zum sekundären Amin reduzieren lässt. Letzteres ist ein geeigneter Reaktionspartner für eine nukleophile Substitution mit Alkyl- oder Allylhalogeniden, welche mit ihm in Dimethylformamid (mit Kaliumcarbonat zum Abfangen der bei der Reaktion freigesetzten Halogenwasserstoffe) die entsprechenden N-6-alkylierten bzw. -allylierten Produkte bilden.[75]
Reaktionen
BearbeitenEine präparativ bedeutende Reaktion der Lysergamide ist die Reduktion der Δ9,10-Doppelbindung, die einen wichtigen Schritt in der Synthese von Ergolin-Arzneistoffen wie Cabergolin, Dihydroergotamin, Dihydroergocryptin und Dihydroergotoxin darstellt.[95] Hierbei entsteht am C-10 ein neues Stereozentrum. Durch katalytische Hydrierung von Lysergsäure und ihren Derivaten wird nur das Stereoisomer mit trans-ständigen Ringen C und D gebildet (bei Isolysergsäure und deren Derivaten dagegen das cis-Isomer), was einen frühen Hinweis auf die jeweilige Stellung der abschirmenden Carbonsäuregruppe bei Lysergsäure und Isolysergsäure gab.[112]
Nach photochemischer Anregung der Doppelbindung kann es in wässriger Lösung auch zur protisch katalysierten Addition von Wasser kommen, bei der sich die Hydroxylgruppe anschließend am C-10 befindet, wobei auch hier das trans-Isomer begünstigt ist.[95]

Insbesondere in Gegenwart katalytischer Mengen Base kommt es leicht zur Epimerisierung am C-8. Der Grund hierfür liegt in der benachbarten Δ9,10-Doppelbindung: (8R)- (Lysergsäure/-amide) und (8S)-Stereoisomer (Isolysergsäure/-amide) können sich über das Δ8,9-Tautomer als Zwischenprodukt ineinander umwandeln. Aus biologischem Material extrahierte Ergopeptine enthalten daher immer auch zu einem gewissen Anteil die Isoformen. Bei Ergopeptamen ist das dagegen nicht der Fall, da diese sich in Gegenwart von Basen rasch zersetzen.[95]
Weitere Reaktionen von Lysergamiden, die in der Literatur beschrieben werden, beinhalten die salzsaure Ringöffnung von 1,2-Dimethylaziridinen in Form ihrer Amide mit Lysergsäure,[113] die vollständige alkalische Hydrolyse von Ergopeptinen sowie die Methanolyse von Ergopeptamen.[95]
Analytik
Bearbeiten
Farbreaktionen können sowohl zum qualitativen als auch zum quantitativen Nachweis von Lysergamiden eingesetzt werden. Zur Anfärbung auf den Trägerplatten nach Dünnschichtchromatographie dient meist das Dragendorff-Reagenz. Hohe Empfindlichkeit bei der photometrischen Bestimmung der Konzentration von Indolverbindungen besitzt die – zum Nachweis der Mutterkornalkaloide entwickelte – Van-Urk-Reaktion. Weitere chemische Methoden sind in der Literatur beschrieben, haben in der heutigen Praxis aber keine große Bedeutung.[114]
Toxikologische Analysen stützen sich hauptsächlich auf instrumentelle Verfahren, insbesondere auf Infrarot- und Kernspinresonanzspektroskopie sowie gekoppelte chromatographische und massenspektrometrische Verfahren (LC/MS und GC/MS).[78][115][116]
Zur Strukturaufklärung können Lysergamide entweder – wie bei ihrer klassischen Synthese aus Ergopeptinen – durch Hydrazinolyse oder durch Reduktion mit Lithiumalanat gespalten werden, zur weiteren Analyse von Peptidteilen können anschließend Standardmethoden wie der Edman-Abbau zum Einsatz kommen. Die Analyse der Fragmente erfolgt dann mittels geeigneter instrumenteller Verfahren.[117]
Regulierung auf Staats- und EU-Ebene
BearbeitenLebens- und Futtermittel
BearbeitenMit der EU-Verordnung 2021/1399 sind mit Wirkung zum 1. Januar 2022 Höchstgehalte von Mutterkornalkaloiden in Getreideerzeugnissen festgelegt. So dürfen bei Abgabe an den Endabnehmer bzw. Verbraucher beispielsweise Roggenmehl und -körner nicht mehr als 500 μg/kg enthalten. Ab 1. Juli 2024 gilt ein abgesenkter Höchstwert von 250 μg/kg. Die Gehalte sind definiert als Summe aus Ergotamin, Ergometrin, Ergosin, Ergocristin, Ergocryptin und Ergocornin und deren Epimeren.[118]
Psychoaktive Substanzen
Bearbeiten
Als LSD in den 60er Jahren zunehmend auch in der breiteren amerikanischen Bevölkerung – insbesondere auch in der Gegenkultur, die sich unter dem Eindruck des Vietnamkrieges herausbildete – als Droge konsumiert wurde, schränkten viele westliche Staaten seit Mitte des Jahrzehnts Herstellung, Inverkehrbringen, Handel und Besitz psychotroper Substanzen rechtlich stark ein. LSD selbst ist seit 1970 in Anhang I des Controlled Substances Act gelistet und unterliegt damit in den USA strengster Regulierung.[32] International fällt es unter die Konvention über psychotrope Substanzen, wodurch es automatisch auch vom österreichischen Suchtmittelgesetz (SMG) erfasst wird. In der Bundesrepublik Deutschland wurde es 1967 mit der vierten Betäubungsmittel-Gleichstellungsverordnung[119] den betäubungsmittelrechtlichen Vorschriften des Opiumgesetzes unterstellt und ist bis heute nicht verkehrsfähig gem. Anlage I zum Betäubungsmittelgesetz (BtMG). Um seine unerlaubte Herstellung zu unterbinden, unterstehen neben Lysergsäure selbst auch die Mutterkornalkaloide Ergometrin und Ergotamin aufgrund ihrer Einstufung als Grundstoffe der Kategorie 1 in der Verordnung (EG) Nr. 111/2005 in Deutschland dem Grundstoffüberwachungsgesetz (GÜG),[120] in der Schweiz zu gleichem Zwecke als Vorläuferstoffe gem. Verzeichnis f der Schweizer Betäubungsmittelverzeichnisverordnung (BetmVV-EDI) dem Betäubungsmittelgesetz (BetmG).[121]
Darüber hinaus unterliegen in Deutschland seit 2016 alle Substanzen den Beschränkungen des Neue-psychoaktive-Stoffe-Gesetzes (NpSG), die gem. § 7 NpSG mit Zustimmung des Bundesrats entspr. Art. 80 Abs. 2 GG durch Verordnung des Bundesgesundheitsministeriums in die Anlage des NpSG eingetragen worden sind. Dort werden zwei Kategorien vom Tryptamin abgeleiteter Verbindungen unterschieden, wobei LSD-Analoga in der Kategorie der Δ9,10-Ergolene erscheinen.[122][123] Das NpSG erfasst jedoch nicht alle Δ9,10-Ergolene schlechthin, sondern gem. § 2 NpSG nur solche, deren molare Masse 500 u nicht überschreitet und die an Position 8 eine Amidgruppe sowie an weiteren bestimmten Positionen der so definierten Lysergamid-Grundstruktur solche Substituenten besitzen, welche in der Anlage des Gesetzes gelistet sind.[122] Eine Unterscheidung nach Konfigurationsisomeren – obwohl pharmakologisch bedeutsam[6] – findet im Rahmen des NpSG nicht statt.[122] Auf Stoffe, die Arzneimittel im Sinne des Arzneimittelgesetzes (AMG) oder Betäubungsmittel im Sinne des BtMG sind, ist das NpSG nicht anwendbar, § 1 Abs. 2 NpSG.
| R1 (am Indol-N) | R2 (am Amin-N) | R3/R4 (am Amid-N) |
|---|---|---|
|
|
|
Das österreichische Recht verfolgt einen etwas anderen Ansatz, hier werden neue psychoaktive Substanzen nicht über ihre chemische Struktur definiert, sondern gem. § 1 NPSG über ihre Fähigkeit, bei ihrer Anwendung im menschlichen Körper eine psychoaktive Wirkung herbeizuführen.[124] Allerdings eröffnet § 3 NPSG dem Bundesgesundheitsminister die Möglichkeit, neue psychoaktive Substanzen mit Verordnung zu bezeichnen. Dabei dürfen – sofern dies besser geeignet erscheint, der Verbreitung psychoaktiver Substanzen vorzubeugen – auch ganze chemische Substanzklassen definiert werden, selbst wenn davon Substanzen mit erfasst werden, deren psychotrope Potenz gering oder nicht vorhanden ist, oder die dem SMG unterliegen. Die Anlage II zur einschlägigen Neue-Psychoaktive-Substanzen-Verordnung (NPSV) benennt pauschal sowohl „[j]ede Verbindung, die von einer Phenethylamin-Grundstruktur abgeleitet werden kann, auch wenn sie ein heterocyclisches oder polycyclisches Ringsystem […] an Stelle der Phenylstruktur oder eine oder mehrere der chemischen Strukturen gemäß § 1 Abs. 2 als Substituent(en) aufweist“ als auch „Tryptamin sowie jede Verbindung, die von dieser chemischen Grundstruktur abgeleitet werden kann, auch wenn sie eine oder mehrere der chemischen Strukturen gemäß § 1 Abs. 2 als Substituent(en) aufweist“, wobei es sich bei Substituenten gemäß § 1 Abs. 2 NPSV um „Aldehyde, Alkane, Alkene, Alkohole, Alkoxide, Alkyle, Alkylhalide, Alkyne, Amide, Amine, Benzyle, Carboxylate, Ester, Ether, Halogenide, Isocyanate, Ketone, Nitrile, Nitroxide, Phenole, Phenyle, Selenoalkyle, Selenoester, Selenole, Thioalkyle, Thiocyanate, Thioester, Thioketone, Thiole, Thiophenole sowie alle chemisch möglichen auch substituierten Ringverbindungen (wie insbesondere Cyclopropyl-, Cyclobutyl-, Benzocyclobutyl- oder Adamantan-Ringstrukturen) und substituierten Hetero-Ringverbindungen (wie insbesondere Indazol-, Pyrazolopyridin-, Azaindol-, Tetrahydro-Naphthyridin-Verbindungen) sowie deren isomere Ringstrukturen“ handeln kann.[125] Nicht anwendbar ist das NPSG auf Stoffe, die nach Maßgabe der arzneimittel-, apotheken- oder arzneiwareneinfuhrrechtlichen Vorschriften in Verkehr gebracht werden dürfen, § 2 NPSG.
In der Schweiz wird vom Eidgenössischen Departement des Innern über die BetmVV-EDI bestimmt, welche Substanzen der Betäubungsmittelkontrolle nach dem BetmG unterliegen. Dort finden sich in Verzeichnis e als „Rohmaterialien und Erzeugnisse mit vermuteter betäubungsmittelähnlicher Wirkung“ mehrere einzelne LSD-Analoga sowie alle Lysergamide, sofern sie
- am Indol-N unsubstituiert oder mit einer beliebigen Alkyl- oder Carbonylgruppe substituiert sind,
- und zusätzlich am Amid-N unsubstituiert oder in beliebigem Umfang mit Alkyl-, Alkenyl-, Alkoxyalkyl- oder Hydroxyalkylgruppen substituiert sind,
- sowie zusätzlich am N-6 mit einer beliebigen Alkyl- oder Alkenylgruppe substituiert sind.[121]
Die Verwendung von LSD und Analoga zu wissenschaftlichen Zwecken ist auch in Deutschland, Österreich und der Schweiz nicht verboten (§ 3 Abs. 2 NpSG) bzw. genehmigungsfähig (§ 3 Abs. 2 BtMG, § 6 Abs. 1 Nr. 2 SMG, Art. 8 Abs. 5, 14 Abs. 2 BetmG).
Weblinks
BearbeitenLiteratur
BearbeitenNicht mehr ganz aktueller, aber sehr umfassender Überblick über Mutterkornalkaloide und ihre Derivate:
- Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: the genus Claviceps. CRC Press, 1999, ISBN 978-90-5702-375-0.
Neuere Einzelarbeiten zur Struktur-Wirkungsbeziehung von Lysergamiden:
- David E. Nichols, Stewart Frescas, Danuta Marona-Lewicka, Deborah M. Kurrasch-Orbaugh: Lysergamides of Isomeric 2,4-Dimethylazetidines Map the Binding Orientation of the Diethylamide Moiety in the Potent Hallucinogenic Agent N,N-Diethyllysergamide (LSD). In: Journal of Medicinal Chemistry, 2002, Band 45, Nr. 19, S. 4344–4349, doi:10.1021/jm020153s.
- Daniel Wacker, Sheng Wang, John D. McCorvy, Robin M. Betz, A. J. Venkatakrishnan, Anat Levit, Katherine Lansu, Zachary L. Schools, Tao Che, David E. Nichols, Brian k. Shoichet, Ron O. Dror, Bryan L. Roth: Crystal Structure of an LSD-Bound Human Serotonin Receptor. In: Cell, 2017, Band 168, S. 377–389, doi:10.1016/j.cell.2016.12.033.
- John D. McCorvy, Daniel Wacker, Sheng Wang, Bemnat Agegnehu, Jing Liu, Katherine Lansu, Alexandra R. Tribo, Reid H. J. Olsen, Tao Che, Jian Jin, Bryan L. Roth: Structural determinants of 5-HT2B receptor activation and biased agonism. In: Nature Structural & Molecular Biology, 2018, Band 25, S. 787–796, doi:10.1038/s41594-018-0116-7.
- Kuglae Kim, Tao Che, Ouliana Panova, Jeffrey F. DiBerto, Jiankun Lyu, Brian E. Krumm, Daniel Wacker, Michael J. Robertson, Alpay B. Seven, David E. Nichols, Brian K. Shoichet, Georgios Skiniotis, Bryan L. Roth: Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor. In: Cell, 2020, Band 182, Nr. 6, S. 1574–1588.e19, doi:10.1016/j.cell.2020.08.024.
- Youwen Zhuang, Peiyu Xu, Chunyou Mao, Lei Wang, Brian Krumm, X. Edward Zhou, Sijie Huang, Heng Liu, Xi Cheng, Xi-Ping Huang, Dan-Dan Shen, Tinghai Xu, Yong-Feng Liu, Yue Wang, Jia Guo, Yi Jiang, Hualiang Jiang, Karsten Melcher, Bryan L. Roth, Yan Zhang, Cheng Zhang, H. Eric Xu: Structural insights into the human D1 and D2 dopamine receptor signaling complexes. In: Cell, 2021, Band 184, Nr. 4, S. 931–942.e18, doi:10.1016/j.cell.2021.01.027.
Aktuelle Einzelarbeiten von Brandt et al. zur analytischen und pharmakologischen Charakterisierung von LSD-Analoga:
- Simon D. Brandt, Pierce V. Kavanagh, Folker Westphal, Alexander Stratford, Simon P. Elliott, Khoa Hoang, Jason Wallach, Adam L. Halberstadt: Return of the lysergamides. Part I: Analytical and behavioural characterization of 1-propionyl-d-lysergic acid diethylamide (1P-LSD). In: Drug Testing and Analysis, 2016, Band 8, Nr. 9, S. 891–902, doi:10.1002/dta.1884.
- Simon D. Brandt, Pierce V. Kavanagh, Folker Westphal, Simon P. Elliott, Jason Wallach, Tristan Colestock, Timothy E. Burrow, Stephen J. Chapman, Alexander Stratford, David E. Nichols, Adam L. Halberstadt: Return of the lysergamides. Part II: Analytical and behavioural characterization of N6-allyl-6-norlysergic acid diethylamide (AL-LAD) and (2’S,4’S)-lysergic acid 2,4-dimethylazetidide (LSZ). In: Drug Testing and Analysis, 2017, Band 9, Nr. 1, S. 38–50, doi:10.1002/dta.1985.
- Simon D. Brandt, Pierce V. Kavanagh, Folker Westphal, Simon P. Elliott, Jason Wallach, Alexander Stratford, David E. Nichols, Adam L. Halberstadt: Return of the lysergamides. Part III: Analytical characterization of N6-ethyl-6-norlysergic acid diethylamide (ETH-LAD) and 1-propionyl ETH-LAD (1P–ETH-LAD). In: Drug Testing and Analysis, 2017, Band 9, Nr. 10, S. 1641–1649, doi:10.1002/dta.2196.
- Simon D. Brandt, Pierce V. Kavanagh, Brendan Twamley, Folker Westphal, Simon P. Elliott, Jason Wallach, Alexander Stratford, Landon M. Klein, John D. McCorvy, David E. Nichols, Adam L. Halberstadt: Return of the lysergamides. Part IV: Analytical and pharmacological characterization of lysergic acid morpholide (LSM-775). In: Drug Testing and Analysis, 2018, Band 10, Nr. 2, S. 310–322, doi:10.1002/dta.2222.
- Simon D. Brandt, Pierce V. Kavanagh, Folker Westphal, Alexander Stratford, Simon P. Elliott, Geraldine Dowling, Jason Wallach, Adam L. Halberstadt: Return of the lysergamides. Part V: Analytical and behavioural characterization of 1-butanoyl-d-lysergic acid diethylamide (1B-LSD). In: Drug Testing and Analysis, 2019, Band 11, Nr. 8, S. 1122–1133, doi:10.1002/dta.2613.
- Simon D. Brandt, Pierce V. Kavanagh, Folker Westphal, Alexander Stratford, Anna U. Odland, Adam K. Klein, Geraldine Dowling, Nicola M. Dempster, Jason Wallach, Torsten Passie, Adam L. Halberstadt: Return of the lysergamides. Part VI: Analytical and behavioural characterization of 1-cyclopropanoyl-d-lysergic acid diethylamide (1CP-LSD). In: Drug Testing and Analysis, 2020, Band 12, Nr. 6, S. 812–826, doi:10.1002/dta.2789.
- Simon D. Brandt, Pierce V. Kavanagh, Folker Westphal, Alexander Stratford, Simon P. Elliott, Geraldine Dowling, Adam L. Halberstadt: Analytical profile of N-ethyl-N-cyclopropyl lysergamide (ECPLA), an isomer of lysergic acid 2,4-dimethylazetidide (LSZ). In: Drug Testing and Analysis. 12, 2020, S. 1514, doi:10.1002/dta.2911.
- Simon D. Brandt, Pierce V. Kavanagh, Folker Westphal, Alexander Stratford, Peter Blanckaert, Geraldine Dowling, Matthias Grill, Hannes M. Schwelm, Volker Auwärter, Stephen J. Chapman: Separating the wheat from the chaff: Observations on the analysis of lysergamides LSD, MIPLA, and LAMPA. In: Drug Testing and Analysis, 2021, Early View, doi:10.1002/dta.3103.
- Simon D. Brandt, Pierce V. Kavanagh, Folker Westphal, Benedikt Pulver, Kathleen Morton, Alexander Stratford, Geraldine Dowling, Adam L. Halberstadt: Return of the lysergamides. Part VII: Analytical and behavioural characterization of 1-valeroyl-D-lysergic acid diethylamide (1V-LSD). In: Drug Testing and Analysis 14, 2022, S. 733, doi:10.1002/dta.3205.
- Simon D. Brandt, Pierce V. Kavanagh, Folker Westphal, Benedikt Pulver, Hannes M. Schwelm, Kyla Whitelock, Alexander Stratford, Volker Auwärter, Adam L. Halberstadt: Analytical profile, in vitro metabolism and behavioral properties of the lysergamide 1P‐AL‐LAD. In: Drug Testing and Analysis. 14, 2022, S. 1503, doi:10.1002/dta.3281.
- Pierce V. Kavanagh, Folker Westphal, Benedikt Pulver, Hannes M. Schwelm, Alexander Stratford, Volker Auwärter, Stephen J. Chapman, Adam L. Halberstadt, Simon D. Brandt: Analytical profile of the lysergamide 1cP‐AL‐LAD and detection of impurities. In: Drug Testing and Analysis. 2022 doi:10.1002/dta.3397.
Anmerkungen
Bearbeiten- ↑ Für eine Darstellung der Phosphoinositidkaskade siehe bspw. Jeremy M. Berg, John L. Tymoczko, Lubert Stryer: Stryer Biochemie. Springer-Verlag, Berlin/Heidelberg 2013, ISBN 978-3-8274-2988-9, S. 411–412.
- ↑ Für eine Darstellung der Drug-Discrimination-Methode siehe Richard A. Glennon: Classical Hallucinogens: An Introductory Overview. In: Geraline C. Lin, Richard A. Glennon (Hrsg.): Hallucinogens: An Update. NIDA Research Monograph 146. National Institute on Drug Abuse, 1994, S. 9 f.
Einzelnachweise
Bearbeiten- ↑ a b Fabrizio Schifano, Laura Orsolini, Duccio Papanti, John Corkery: NPS: Medical Consequences Associated with Their Intake. In: Michael H. Baumann, Richard A. Glennon, Jenny L. Wiley: Neuropharmacology of New Psychoactive Substances (NPS). Springer, E-Book 2016, ISBN 978-3-319-52444-3, S. 364.
- ↑ Christopher L. Schardl, Daniel G. Panaccione, Paul Tudzynski: Ergot Alkaloids – Biology and Molecular Biology. In: The Alkaloids: Chemistry and Biology – Volume 63. Elsevier 2006, doi:10.1016/S1099-4831(06)63002-2, S. 67.
- ↑ Christopher L. Schardl, Daniel G. Panaccione, Paul Tudzynski: Ergot Alkaloids – Biology and Molecular Biology. In: The Alkaloids: Chemistry and Biology – Volume 63. Elsevier 2006, doi:10.1016/S1099-4831(06)63002-2, S. 46–47.
- ↑ S. Pakhomova, J. Ondráček, M. Hušak, B. Kratochvíl: Conformation of ergopeptam and ergopeptine alkaloids (ergocristam and ergocristine). In: Zeitschrift für Kristallographie, 1997, Band 212, Nr. 8, S. 593–600, doi:10.1524/zkri.1997.212.8.593.
- ↑ Christopher L. Schardl, Daniel G. Panaccione, Paul Tudzynski: Ergot Alkaloids – Biology and Molecular Biology. In: The Alkaloids: Chemistry and Biology – Volume 63. Elsevier 2006, doi:10.1016/S1099-4831(06)63002-2, S. 49.
- ↑ a b c d Robert C. Pfaff, Xuemei Huang, Danuta Marona-Lewicka, Robert Oberlender, David E. Nichols: Lysergamides Revisited. In: Geraline C. Lin, Richard A. Glennon (Hrsg.): Hallucinogens: An Update. NIDA Research Monograph 146. National Institute on Drug Abuse, 1994, S. 53–54.
- ↑ Adam L. Halberstadt, Landon M. Klein, Muhammad Chatha, Laura B. Valenzuela, Alexander Stratford, Jason Wallach, David E. Nichols, Simon D. Brandt: Pharmacological characterization of the LSD analog N-ethyl-N-cyclopropyl lysergamide (ECPLA). In: Psychopharmacology, 2019, Band 236, S. 799–808, doi:10.1007/s00213-018-5055-9.
- ↑ Harold A. Abramson: Lysergic Acid Diethylamide (LSD-25): XXXI. Comparison by Questionnaire of Psychotomimetic Activity of Congeners on Normal Subjects and Drug Addicts. In: British Journal of Psychiatry, 1960, Band 106, S. 1120–1123, doi:10.1192/bjp.106.444.1120.
- ↑ a b Irmtraut Sahmland: Ergotismus. In: Werner E. Gerabek, Bernhard D. Haage, Gundolf Keil, Wolfgang Wegner (Hrsg.): Enzyklopädie Medizingeschichte. De Gruyter, Berlin / New York 2007, ISBN 978-3-11-019703-7, S. 367–368.
- ↑ Charles Tanret: Sur la présence d’un nouvel alcaloïde, l’ergotinine, dans le seigle ergoté. In: Comptes rendus hebdomadaires des séances de l’Académie des sciences, 1875, Band 81, S. 896–897. Online archiviert auf Gallica.
- ↑ Charles Tanret: De l’ergotinine. In: Annales de chimie et de physique, 1879, Band 17, S. 493–512. Online archiviert auf Gallica.
- ↑ a b Léo Marion: The Indole Alkaloids. In: R. H. F. Manske, H. L. Holmes (Hrsg.): The Alkaloids: Chemistry and Physiology – Volume II. Academic Press, New York 1952, S. 375–378.
- ↑ Laurence Lestel: Itinéraires de chimistes. EDP Sciences, 2007, doi:10.1051/978-2-7598-0315-6.c085, S. 502.
- ↑ G. Barger, F. H. Carr, H. H. Dale: An Active Alkaloid From Ergot. In: The British Medical Journal, 1906, Band 2, Nr. 2399 (Dec. 22, 1906), S. 1792. Archiviert auf JSTOR.
- ↑ George Barger, Francis Howard Carr: XXXVII.—The alkaloids of ergot. In: Journal of the Chemical Society, Transactions, 1907, Band 91, S. 337–353, doi:10.1039/CT9079100337.
- ↑ F. Kraft: Ueber das Mutterkorn. In: Archiv der Pharmazie, 1906, Band 244, Nr. 4–5, S. 336–359, doi:10.1002/ardp.19062440411.
- ↑ Arthur Stoll, Albert Hofmann: Die Alkaloide der Ergotoxingruppe: Ergocristin, Ergokryptin und Ergocornin. 7. Mitteilung über Mutterkornalkaloide. In: Helvetica Chimica Acta, 1943, Band 26, Nr. 5, S. 1570–1601, doi:10.1002/hlca.19430260522.
- ↑ Arthur Stoll, Albert Hofmann: The Ergot Alkaloids. In: R. H. F. Manske (Hrsg.): The Alkaloids: Chemistry and Physiology – Volume VIII. Academic Press, New York / London 1965, S. 727.
- ↑ Arthur Stoll: Zur Kenntnis der Mutterkornalkaloide. In: Verhandlungen der Schweizerischen Naturforschenden Gesellschaft, 1920, Band 101, S. 190–191. Online archiviert auf e-periodica.ch (ETH Zürich).
- ↑ K. Spiro, A. Stoll: Über die wirksamen Substanzen des Mutterkorns. In: Verhandlungen der Schweizerischen Naturforschenden Gesellschaft, 1920, Band 101, S. 235–236. Online archiviert auf e-periodica.ch (ETH Zürich).
- ↑ Michael Radcliffe Lee: The history of ergot of rye (Claviceps purpurea) II: 1900–1940. In: Journal of the Royal College of Physicians of Edinburgh, 2009, Band 39, Nr. 4, S. 365–369, doi:10.4997/JRCPE.2009.416.
- ↑ a b Arthur Stoll, Albert Hofmann: Partialsynthese von Alkaloiden vom Typus des Ergobasins. 6. Mitteilung über Mutterkornalkaloide. In: Helvetica Chimica Acta, 1943, Band 26, Nr. 3, S. 944–965, doi:10.1002/hlca.19430260326.
- ↑ a b c Michael Radcliffe Lee: The history of ergot of rye (Claviceps purpurea) III: 1940–80. In: Journal of the Royal College of Physicians of Edinburgh, 2010, Band 40, Nr. 1, S. 77–80, doi:10.4997/jrcpe.2010.115.
- ↑ Eintrag zu Methylergometrin. In: Römpp Online. Georg Thieme Verlag, abgerufen am 15. Juli 2019.
- ↑ a b Albert Sjoerdsma, Michael G. Palfreyman: History of serotonin and serotonin disorders. In: Annals of the New York Academy of Sciences, 1990, Band 600, S. 1–8, doi:10.1111/j.1749-6632.1990.tb16869.x.
- ↑ J. H. Gaddum, C. O. Hebb, Ann Silver, A. A. B. Swan: 5-Hydroxytryptamine. Pharmacological action and destruction in perfused lungs. In: Quarterly Journal of Experimental Physiology and Cognate Medical Sciences, 1953, Band 38, Nr. 4, S. 255–262, doi:10.1113/expphysiol.1953.sp001037.
- ↑ D. W. Woolley, E. Shaw: A biochemical and pharmacological suggestion about certain mental disorders. In: Proceedings of the National Academy of Sciences of the United States of America, 1954, Band 40, Nr. 4, S. 228–231, doi:10.1073/pnas.40.4.228.
- ↑ E. Shaw, D. W. Woolley: Some Serotoninlike Activities of Lysergic Acid Diethylamide. In: Science, 1956, Band 124, Nr. 3212, S. 121–122, doi:10.1126/science.124.3212.121.
- ↑ A. Cerletti, E. Rothlin: Role of 5-Hydroxytryptamine in Mental Diseases and its Antagonism to Lysergic Acid Derivatives. In: Nature, 1955, Band 176, Nr. 4486, S. 785–786, doi:10.1038/176785a0.
- ↑ David E. Nichols: Dark Classics in Chemical Neuroscience: Lysergic Acid Diethylamide (LSD). In: ACS Chemical Neuroscience, 2018, Band 9, Nr. 10, S. 2331–2343, doi:10.1021/acschemneuro.8b00043.
- ↑ Nadine Weidman: Matthew Oram. The Trials of Psychedelic Therapy: LSD Psychotherapy in America (Buchbesprechung). In: Isis, 2020, Band 111, Nr. 1, doi:10.1086/707842. Online auf der Website der University of Chicago (PDF) abgerufen am 1. August 2021.
- ↑ a b Matthew Oram: Prohibited or regulated? LSD psychotherapy and the United States Food and Drug Administration. In: History of Psychiatry, 2016, Band 27, Nr. 3, S. 290–306, doi:10.1177/0957154X16648822.
- ↑ a b c d e f g h Daniel Wacker, Sheng Wang, John D. McCorvy, Robin M. Betz, A. J. Venkatakrishnan, Anat Levit, Katherine Lansu, Zachary L. Schools, Tao Che, David E. Nichols, Brian k. Shoichet, Ron O. Dror, Bryan L. Roth: Crystal Structure of an LSD-Bound Human Serotonin Receptor. In: Cell, 2017, Band 168, S. 377–389, doi:10.1016/j.cell.2016.12.033.
- ↑ Weltgesundheitsorganisation: WHO Model Lists of Essential Medicines, 21. Auflage, 2019, S. 47.
- ↑ Christof Schaefer, Horst Spielmann, Klaus Vetter (Hrsg.): Arzneiverordnung in Schwangerschaft und Stillzeit. Urban & Fischer / Elsevier, München 2006, ISBN 978-3-437-21332-8, S. 358.
- ↑ Pschyrembel Online: Ergometrin. Version von 01.2021, abgerufen am 17. September 2021.
- ↑ Ulrich Schwabe: Gynäkologika. In: Ulrich Schwabe, Dieter Paffrath (Hrsg.): Arzneiverordnungs-Report 2012. Springer, Berlin/Heidelberg 2012, ISBN 978-3-642-29242-2 (E-Book), S. 630–631.
- ↑ R. W. Griffith, J. Grauwiler, Ch. Hodel, K. H. Leist, B. Matter: Toxicologic Considerations. In: B. Herde, H. O. Schild (Hrsg.): Ergot Alkaloids and Related Compounds. Springer-Verlag, Berlin / Heidelberg / New York 1978, ISBN 978-3-642-66777-0, S. 827–828.
- ↑ P. J. Koehler, P. C. Tfelt-Hansen: History of Methysergide in Migraine. In: Cephalalgia, 2008, Band 28, Nr. 11, S. 1126–1135, doi:10.1111/j.1468-2982.2008.01648.x
- ↑ DrugPatentWatch: Details for New Drug Application (NDA): 012516, abgerufen am 2. August 2021.
- ↑ Matthias Karst, John H. Halpern, Michael Bernateck, Torsten Passie: The non-hallucinogen 2-bromo-lysergic acid diethylamide as preventative treatment for cluster headache: An open, non-randomized case series. In: Cephalalgia, 2010, Band 30, Nr. 9, S. 1140–1144, doi:10.1177/0333102410363490.
- ↑ Julia Schaufler, Carsten Telschow, Jana Weiß: Ergänzende statistische Übersicht. In: Ulrich Schwabe, Dieter Paffrath (Hrsg.): Arzneiverordnungs-Report 2014. Springer, Berlin/Heidelberg 2014, ISBN 978-3-662-43487-1 (E-Book), S. 1127.
- ↑ Ulrich Schwabe: Hypophysen- und Hypothalamushormone. In: Ulrich Schwabe, Dieter Paffrath (Hrsg.): Arzneiverordnungs-Report 2020. Springer, Berlin/Heidelberg 2020, ISBN 978-3-662-62168-4 (E-Book), S. 590.
- ↑ a b Melanie Schröder, Carsten Telschow, Jana Weiss: Ergänzende statistische Übersicht. In: Ulrich Schwabe, Dieter Paffrath, Wolf-Dieter Ludwig, Jürgen Klauber (Hrsg.): Arzneiverordnungs-Report 2018. Springer, Berlin/Heidelberg 2018, ISBN 978-3-662-57386-0 (E-Book), S. 855.
- ↑ Julia Schaufler, Carsten Telschow, Jana Weiß: Ergänzende statistische Übersicht. In: Ulrich Schwabe, Dieter Paffrath (Hrsg.): Arzneiverordnungs-Report 2014. Springer, Berlin/Heidelberg 2014, ISBN 978-3-662-43487-1 (E-Book), S. 1155.
- ↑ Valentina Coca, Helmut Schröder: Ergänzende statistische Übersicht. In: Ulrich Schwabe, Dieter Paffrath (Hrsg.): Arzneiverordnungs-Report 2012. Springer, Berlin/Heidelberg 2012, ISBN 978-3-642-29242-2 (E-Book), S. 1020.
- ↑ Heinz Pertz, Eckart Eich: Ergot alkaloids and their derivatives as ligands for serotoninergic, dopaminergic and adrenergic receptors. In: Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: the genus Claviceps. CRC Press, 1999, ISBN 978-90-5702-375-0, S. 411–412.
- ↑ Wesley K. Kroeze, Maria F. Sassano, Xi-Ping Huang, Katherine Lansu, John D. McCorvy, Patrick M. Giguère, Noah Sciaky, Bryan L. Roth: PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. In: Nature Structural & Molecular Biology, 2015, Band 22, S. 362–369, doi:10.1038/nsmb.3014.
- ↑ a b Matthias E. Liechti: Modern Clinical Research on LSD. In: Neuropsychopharmacology, 2017, Band 42, S. 2114–2127, doi:10.1038/npp.2017.86.
- ↑ a b Vargas MV, Dunlap LE, Dong C et al.: Psychedelics promote neuroplasticity through the activation of intracellular 5-HT2A receptors. In: Science. 379. Jahrgang, Nr. 6633, 2023, S. 700–706, doi:10.1126/science.adf0435, PMID 36795823.
- ↑ David E. Nichols, Eric L. Barker: Psychedelics. In: Pharmacological Reviews, 2016, Band 68, Nr. 2, S. 264–355, doi:10.1124/pr.115.011478.
- ↑ a b Wallach J, Cao AB, Calkins MM, et al.: Identification of 5-HT 2A Receptor Signaling Pathways Responsible for Psychedelic Potential. In: bioRxiv. Vorschauversion. Jahrgang, 2023, doi:10.1101/2023.07.29.551106, PMID 37577474, PMC 10418054 (freier Volltext).
- ↑ Ramona M. Rodriguiz, Vineet Nadkarni, Christopher R. Means, Vladimir M. Pogorelov, Yi-Ting Chiu, Bryan L. Roth, William C. Wetsel: LSD-stimulated behaviors in mice require β-arrestin 2 but not β-arrestin 1. In: Scientific Reports, 2021, Band 11, 17690, doi:10.1038/s41598-021-96736-3.
- ↑ Lewis V, Bonniwell EM, Lanham JK et al.: A non-hallucinogenic LSD analog with therapeutic potential for mood disorders. In: Cell Rep. 42. Jahrgang, Nr. 3, 2023, S. 112203, doi:10.1016/j.celrep.2023.112203, PMID 36884348, PMC 10112881 (freier Volltext).
- ↑ Olson DE: Biochemical Mechanisms Underlying Psychedelic-Induced Neuroplasticity. In: Biochemistry. 61. Jahrgang, Nr. 3, 2022, S. 127–136, doi:10.1021/acs.biochem.1c00812, PMID 35060714, PMC 9004607 (freier Volltext).
- ↑ a b Moliner R, Girych M, Brunello CA et al.: Psychedelics promote plasticity by directly binding to BDNF receptor TrkB. In: Nat Neurosci. 26. Jahrgang, Nr. 6, 2023, S. 1032–1041, doi:10.1038/s41593-023-01316-5, PMID 37280397, PMC 10244169 (freier Volltext).
- ↑ Casarotto PC, Girych M, Fred SM et al.: Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. In: Cell. 184. Jahrgang, Nr. 5, 2021, S. 1299–1313.e19, doi:10.1016/j.cell.2021.01.034, PMID 33606976, PMC 7938888 (freier Volltext).
- ↑ a b Daniel Wacker, Chong Wang, Vsevolod Katritch, Gye Won Han, Xi-Ping Huang, Eyal Vardy, John D. McCorvy, Yi Jiang, Meihua Chu, Fai Yiu Siu, Wei Liu, H. Eric Xu, Vadim Cherezov, Bryan L. Roth, Raymond C. Stevens: Structural Features for Functional Selectivity at Serotonin Receptors. In: Science, 2013, Band 340, Nr. 6132, S. 615–619, doi:10.1126/science.1232808.
- ↑ a b John D. McCorvy, Daniel Wacker, Sheng Wang, Bemnat Agegnehu, Jing Liu, Katherine Lansu, Alexandra R. Tribo, Reid H. J. Olsen, Tao Che, Jian Jin, Bryan L. Roth: Structural determinants of 5-HT2B receptor activation and biased agonism. In: Nature Structural & Molecular Biology, 2018, Band 25, S. 787–796, doi:10.1038/s41594-018-0116-7.
- ↑ Heinz Pertz, Eckart Eich: Ergot alkaloids and their derivatives as ligands for serotoninergic, dopaminergic and adrenergic receptors. In: Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: the genus Claviceps. CRC Press, 1999, ISBN 978-90-5702-375-0, S. 425–429.
- ↑ Norio Ogawa, Ken-ichi Tanaka, Masato Asanuma: Bromocriptine Markedly Suppresses Levodopa-Induced Abnormal Increase of Dopamine Turnover in the Parkinsonian Striatum. In: Neurochemical Research, 2000, Band 25, Nr. 6, S. 755–758, doi:10.1023/A:1007530720544.
- ↑ Danuta Marona-Lewicka, David E. Nichols: Further evidence that the delayed temporal dopaminergic effects of LSD are mediated by a mechanism different than the first temporal phase of action. In: Pharmacology Biochemistry and Behavior, 2007, Band 87, Nr. 4, S. 453–461, doi:10.1016/j.pbb.2007.06.001.
- ↑ Heinz Pertz, Eckart Eich: Ergot alkaloids and their derivatives as ligands for serotoninergic, dopaminergic and adrenergic receptors. In: Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: the genus Claviceps. CRC Press, 1999, ISBN 978-90-5702-375-0, S. 429–430.
- ↑ Peer C. Tfelt-Hansen: Triptans and ergot alkaloids in the acute treatment of migraine: similarities and differences. In: Expert Review of Neurotherapeutics, 2013, Band 13, Nr. 9, S. 961–963, doi:10.1586/14737175.2013.832851.
- ↑ Val. J. Watts, Cindy P. Lawler, David R. Fox, Kim A. Neve, David E. Nichols, Richard B. Mailman: LSD and structural analogs: pharmacological evaluation at D1 dopamine receptors. In: Psychopharmacology, 1995, Band 118, S. 401–409, doi:10.1007/BF02245940.
- ↑ a b c Adam L. Halberstadt, Muhammad Chatha, Adam K. Klein, John D. McCorvy, Markus R. Meyer, Lea Wagmann, Alexander Stratford, Simon D. Brandt: Pharmacological and biotransformation studies of 1-acyl-substituted derivatives of d-lysergic acid diethylamide (LSD). In: Neuropharmacology, 2020, Band 172, S. 107856, doi:10.1016/j.neuropharm.2019.107856.
- ↑ a b Leigh D. Coney, Larissa J. Maier, Jason A. Ferris, Adam R. Winstock, Monica J. Barratt: Genie in a blotter: A comparative study of LSD and LSD analogues’ effects and user profile. In: Human Psychopharmacology: Clinical and Experimental, 2017, Band 32, Nr. 3, e2599, doi:10.1002/hup.2599.
- ↑ a b Jie Yin, Kuang-Yui M. Chen, Mary J. Clark, Mahdi Hijazi, Punita Kumari, Xiao-Chen Bai, Roger K. Sunahara, Patrick Barth, Daniel M. Rosenbaum: Structure of a D2 dopamine receptor-G-protein complex in a lipid membrane. In: Nature, 2020, Band 584, Nr. 7819, S. 125–129, doi:10.1038/s41586-020-2379-5.
- ↑ a b Youwen Zhuang, Peiyu Xu, Chunyou Mao, Lei Wang, Brian Krumm, X. Edward Zhou, Sijie Huang, Heng Liu, Xi Cheng, Xi-Ping Huang, Dan-Dan Shen, Tinghai Xu, Yong-Feng Liu, Yue Wang, Jia Guo, Yi Jiang, Hualiang Jiang, Karsten Melcher, Bryan L. Roth, Yan Zhang, Cheng Zhang, H. Eric Xu: Structural insights into the human D1 and D2 dopamine receptor signaling complexes. In: Cell, 2021, Band 184, Nr. 4, S. 931–942.e18, doi:10.1016/j.cell.2021.01.027.
- ↑ a b c David E. Nichols, Stewart Frescas, Danuta Marona-Lewicka, Deborah M. Kurrasch-Orbaugh: Lysergamides of Isomeric 2,4-Dimethylazetidines Map the Binding Orientation of the Diethylamide Moiety in the Potent Hallucinogenic Agent N,N-Diethyllysergamide (LSD). In: Journal of Medicinal Chemistry, 2002, Band 45, Nr. 19, S. 4344–4349, doi:10.1021/jm020153s.
- ↑ David E. Nichols, A. Monte, X. Huang, D. Marona-Lewick: Stereoselective pharmacological effects of lysergic acid amides possessing chirality in the amide substituent. In: Behavioural Brain Research. 1995, Band 73, Nr. 1–2, S. 117–119, doi:10.1016/0166-4328(96)00080-0
- ↑ Xuemei Huang, Danuta Marona-Lewicka, Robert C. Pfaff, David E. Nichols: Drug discrimination and receptor binding studies of N-isopropyl lysergamide derivatives. In: Pharmacology Biochemistry and Behavior, 1994, Band 47, Nr. 3, S. 667–673, doi:10.1016/0091-3057(94)90172-4.
- ↑ Kuglae Kim, Tao Che, Ouliana Panova, Jeffrey F. DiBerto, Jiankun Lyu, Brian E. Krumm, Daniel Wacker, Michael J. Robertson, Alpay B. Seven, David E. Nichols, Brian K. Shoichet, Georgios Skiniotis, Bryan L. Roth: Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor. In: Cell, 2020, Band 182, Nr. 6, S. 1574–1588.e19, doi:10.1016/j.cell.2020.08.024.
- ↑ Robert C. Pfaff, Xuemei Huang, Danuta Marona-Lewicka, Robert Oberlender, David E. Nichols: Lysergamides Revisited. In: Geraline C. Lin, Richard A. Glennon (Hrsg.): Hallucinogens: An Update. NIDA Research Monograph 146. National Institute on Drug Abuse, 1994, S. 55.
- ↑ a b c Andrew J. Hoffman, David E. Nichols: Synthesis and LSD-like discriminative stimulus properties in a series of N(6)-alkyl norlysergic acid N,N-diethylamide derivatives. In: Journal of Medicinal Chemistry, 1985, Band 28, Nr. 9, S. 1252–1255, doi:10.1021/jm00147a022.
- ↑ Robert C. Pfaff, Xuemei Huang, Danuta Marona-Lewicka, Robert Oberlender, David E. Nichols: Lysergamides Revisited. In: Geraline C. Lin, Richard A. Glennon (Hrsg.): Hallucinogens: An Update. NIDA Research Monograph 146. National Institute on Drug Abuse, 1994, S. 57.
- ↑ Michael P. Johnson, Richard J. Loncharich, Melvyn Baez, David L. Nelson: Species variations in transmembrane region V of the 5-hydroxytryptamine type 2A receptor alter the structure-activity relationship of certain ergolines and tryptamines. In: Molecular Pharmacology, 1997, Band 45, S. 277–286.
- ↑ a b c d e f Lea Wagmann, Lilian H. J. Richter, Tobias Kehl, Franziska Wack, Madeleine Pettersson Bergstrand, Simon D. Brandt, Alexander Stratford, Hans H. Maurer, Markus R. Meyer: In vitro metabolic fate of nine LSD-based new psychoactive substances and their analytical detectability in different urinary screening procedures. In: Analytical and Bioanalytical Chemistry, 2019, Band 411, S. 4751–4763, doi:10.1007/s00216-018-1558-9.
- ↑ Patrick Vizeli, Isabelle Straumann, Friederike Holze, Yasmin Schmid, Patrick C. Dolder, Matthias E. Liechti: Genetic influence of CYP2D6 on pharmacokinetics and acute subjective effects of LSD in a pooled analysis. In: Scientific Reports, 2021, Band 11, Nr. 1, 10851 doi:10.1038/s41598-021-90343-y.
- ↑ U. Bredberg, G. S. Eyjolfsdottir, L. Paalzow, P. Tfelt-Hansen, V. Tfelt-Hansen: Pharmacokinetics of Methysergide and its Metabolite Methylergometrine in Man. In: European Journal of Clinical Pharmacology, 1986, Band 30, S. 75–77, doi:10.1007/BF00614199.
- ↑ Pieter S. Steyn: Mycotoxins, general view, chemistry and structure. In: Toxicology Letters, 1995, Band 82–83, S. 843–851, doi:10.1016/0378-4274(95)03525-7.
- ↑ M. Peraica, B. Radić, A. Lucić, M. Pavlović: Toxic effects of mycotoxins in humans. In: Bulletin of the World Health Organization, 1999, Band 77, Nr. 9, S. 754–766, PMID 10534900.
- ↑ Mervyn J. Eadie: Convulsive ergotism: epidemics of the serotonin syndrome? In: The Lancet Neurology, 2003, Band 2, Nr. 7, S. 429–434, doi:10.1016/S1474-4422(03)00439-3.
- ↑ Sarah Belser-Ehrlich, Ashley Harper, John Hussey, Robert Hallock: Human and cattle ergotism since 1900: Symptoms, outbreaks, and regulations. In: Toxicology and Industrial Health, 2012, Band 29, Nr. 4, S. 307–316, doi:10.1177/0748233711432570.
- ↑ S. Srisuma, E. J. Lavonas, W. Wananukul: Ergotism and factitious hypotension associated with interaction of ergotamine with CYP3A4 inhibitors. In: Clinical Toxicology, 2014, Band 52, Nr. 7, S. 674–677, doi:10.3109/15563650.2014.933230.
- ↑ R. W. Griffith, J. Grauwiler, Ch. Hodel, K. H. Leist, B. Matter: Toxicologic Considerations. In: B. Herde, H. O. Schild (Hrsg.): Ergot Alkaloids and Related Compounds. Springer-Verlag, Berlin / Heidelberg / New York 1978, ISBN 978-3-642-66777-0, S. 817.
- ↑ G. Roberto, E. Raschi, C. Piccinni, V. Conti, L. Vignatelli, R. D’Alessandro, F. De Ponti, E. Poluzzi: Adverse cardiovascular events associated with triptans and ergotamines for treatment of migraine: Systematic review of observational studies. In: Cephalalgia, 2015, Band 35, Nr. 2, S. 118–131, doi:10.1177/0333102414550416.
- ↑ Dennis Mulac, Hans-Ulrich Humpf: Cytotoxicity and accumulation of ergot alkaloids in human primary cells. In: Toxicology, 2011, Band 282, Nr. 3, S. 112–121, doi:10.1016/j.tox.2011.01.019.
- ↑ David E. Nichols, Charles S. Grob: Is LSD toxic? In: Forensic Science International, 2018, Band 284, S. 141–145, doi:10.1016/j.forsciint.2018.01.006.
- ↑ Dino Luethi, Matthias E. Liechti: Designer drugs: mechanism of action and adverse effects. In: Archives of Toxicology, 2020, Band 94, S. 1085–1133, doi:10.1007/s00204-020-02693-7.
- ↑ a b c d e Nina Gerhards, Lisa Neubauer, Paul Tudzynski, Shu-Ming Li: Biosynthetic Pathways of Ergot Alkaloids. In: Toxins, 2014, Band 6, Nr. 12, S. 3281–3295, doi:10.3390/toxins6123281.
- ↑ Telmo Correia, Nicolas Grammel, Ingo Ortel, Ullrich Keller, Paul Tudzynski: Molecular Cloning and Analysis of the Ergopeptine Assembly System in the Ergot Fungus Claviceps purpurea. In: Chemistry & Biology, 2003, Band 10, Nr. 12, S. 1281–1292, doi:10.1016/j.chembiol.2003.11.013.
- ↑ Ladislav Cvak: Industrial Production of Ergot Alkaloids. In: Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: The Genus Claviceps. Medicinal and Aromatic Plants – Industrial Profiles. CRC Press, 1999, ISBN 0-203-30419-5, S. 374.
- ↑ J. Rutschmann, P. A. Stadler: Chemical Background. In: B. Herde, H. O. Schild (Hrsg.): Ergot Alkaloids and Related Compounds. Springer-Verlag, Berlin / Heidelberg / New York 1978, ISBN 978-3-642-66777-0, S. 31–32.
- ↑ a b c d e f E. L. Komarova, O. N. Tolkachev: The Chemistry of Peptide Ergot Alkaloids. Part 1. Classification and Chemistry of Ergot Peptides. In: Pharmaceutical Chemistry Journal, 2001, Band 35, S. 504–513, doi:10.1023/A:1014050926916.
- ↑ Ladislav Cvak: Industrial Production of Ergot Alkaloids. In: Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: The Genus Claviceps. Medicinal and Aromatic Plants – Industrial Profiles. CRC Press, 1999, ISBN 0-203-30419-5, S. 377.
- ↑ Edmund C. Kornfeld, E. J. Fornefeld, G. Bruce Kline, Marjorie J. Mann, Dwight E. Morrison, Reuben G. Jones, and Robert B. Woodward: The Total Synthesis of Lysergic Acid. In: Journal of the American Chemical Society, 1956, Band 78, Nr. 13, S. 3087–3114, doi:10.1021/ja01594a039.
- ↑ s. Shinsuke Inuki, Akira Iwata, Shinya Oishi, Nobutaka Fujii, Hiroaki Ohno: Enantioselective Total Synthesis of (+)-Lysergic Acid, (+)-Lysergol, and (+)-Isolysergol by Palladium-Catalyzed Domino Cyclization of Allenes Bearing Amino and Bromoindolyl Groups. In: The Journal of Organic Chemistry, 2011, Band 76, Nr. 7, S. 2072–2083, doi:10.1021/jo102388e, m. w. N.
- ↑ Ladislav Cvak: Industrial Production of Ergot Alkaloids. In: Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: The Genus Claviceps. Medicinal and Aromatic Plants – Industrial Profiles. CRC Press, 1999, ISBN 0-203-30419-5, S. 378.
- ↑ Petr Bulej, Ladislav Cvak: Chemical modifications of ergot alkaloids. In: Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: The Genus Claviceps. Medicinal and Aromatic Plants – Industrial Profiles. CRC Press, 1999, ISBN 0-203-30419-5, S. 201 ff., v. a. 208–209.
- ↑ A. Stoll, A. Hofmann: Die optisch aktiven Hydrazide der Lysergsäure und der Isolysergsäure. (4. Mitteilung über Mutterkornalkaloide). In: Helvetica Chimica Acta, 1943, Band 26, Nr. 3, S. 922–928, doi:10.1002/hlca.19430260324.
- ↑ a b Fatima N. Johnson, Istvan E. Ary, David G. Teiger, Ronald J. Kassel: Emetic Activity of Reduced Lysergamides. In: Journal of Medicinal Chemistry, 1973, Band 16, Nr. 5, S. 532–537, doi:10.1021/jm00263a028.
- ↑ a b William L. Garbrecht: Synthesis of Amides of Lysergic Acid. In: The Journal of Organic Chemistry, 1959, Band 24, Nr. 3, S. 368–372, doi:10.1021/jo01085a024.
- ↑ Petr Bulej, Ladislav Cvak: Chemical modifications of ergot alkaloids. In: Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: The Genus Claviceps. Medicinal and Aromatic Plants – Industrial Profiles. CRC Press, 1999, ISBN 0-203-30419-5, S. 202.
- ↑ F. Troxler, A. Hofmann: Substitutionen am Ringsystem der Lysergsäure II. Alkylierung. 44. Mitteilung über Mutterkornalkaloide. In: Helvetica Chimica Acta, 1957, Band 40, Nr. 6, S. 1721–1732, doi:10.1002/hlca.19570400620.
- ↑ Petr Bulej, Ladislav Cvak: Chemical modifications of ergot alkaloids. In: Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: The Genus Claviceps. Medicinal and Aromatic Plants – Industrial Profiles. CRC Press, 1999, ISBN 0-203-30419-5, S. 203.
- ↑ F. Troxler, A. Hofmann: Substitutionen am Ringsystem der Lysergsäure I. Substitutionen am Indol-Stickstoff. 43. Mitteilung über Mutterkornalkaloide. In: Helvetica Chimica Acta, 1957, Band 40, Nr. 6, S. 1706–1720, doi:10.1002/hlca.19570400619.
- ↑ s. Petr Bulej, Ladislav Cvak: Chemical modifications of ergot alkaloids. In: Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: The Genus Claviceps. Medicinal and Aromatic Plants – Industrial Profiles. CRC Press, 1999, ISBN 0-203-30419-5, S. 202–203, m. w. N.
- ↑ F. Troxler, A. Hofmann: Substitutionen am Ringsystem der Lysergsäure. III. Halogenierung. 45. Mitteilung über Mutterkornalkaloide. In: Helvetica Chimica Acta, 1957, Band 40, Nr. 7, S. 2160–2170, doi:10.1002/hlca.19570400716.
- ↑ Petr Bulej, Ladislav Cvak: Chemical modifications of ergot alkaloids. In: Vladimír Křen, Ladislav Cvak (Hrsg.): Ergot: The Genus Claviceps. Medicinal and Aromatic Plants – Industrial Profiles. CRC Press, 1999, ISBN 0-203-30419-5, S. 204–205.
- ↑ Jie Jack Li: Name Reactions: A Collection of Detailed Reaction Mechanisms. Springer, Berlin/Heidelberg 2006, ISBN 978-3-540-30031-1 (E-Book), S. 608–609.
- ↑ Arthur Stoll, Albert Hofmann: The Ergot Alkaloids. In: R. H. F. Manske (Hrsg.): The Alkaloids: Chemistry and Physiology – Volume VIII. Academic Press, New York / London 1965, S. 738.
- ↑ Robert C. Pfaff, Xuemei Huang, Danuta Marona-Lewicka, Robert Oberlender, David E. Nichols: Lysergamides Revisited. In: Geraline C. Lin, Richard A. Glennon (Hrsg.): Hallucinogens: An Update. NIDA Research Monograph 146. National Institute on Drug Abuse, 1994, S. 59–62.
- ↑ a b E. L. Komarova, O. N. Tolkachev: The Chemistry of Peptide Ergot Alkaloids. Part 2. Analytical Methods for Determining Ergot Alkaloids. In: Pharmaceutical Chemistry Journal, 2001, Band 35, S. 542–549, doi:10.1023/A:1014706301632.
- ↑ Colin Crews: Analysis of Ergot Alkaloids. In: Toxins, 2015, Band 7, Nr. 6, S. 2024–2050, doi:10.3390/toxins7062024.
- ↑ vgl. die im Literaturverzeichnis aufgeführten Arbeiten von Brandt et al.
- ↑ Gerhard Habermehl, Peter E. Hammann, Hans C. Krebs, W. Ternes: Naturstoffchemie. Springer, Berlin/Heidelberg 2008, ISBN 978-3-540-73733-9 (E-Book), S. 204.
- ↑ Verordnung (EU) 2021/1399 der Kommission vom 24. August 2021 zur Änderung der Verordnung (EG) Nr. 1881/2006 hinsichtlich der Höchstgehalte an Mutterkorn-Sklerotien und Ergotalkaloiden in bestimmten Lebensmitteln. Online auf EUR-Lex, abgerufen am 23. Januar 2022.
- ↑ BtMGlV vom 21. Februar 1967. BGBl. I S. 197 – Nr. 10 – Ausgegeben zu Bonn am 24. Februar 1967.
- ↑ Verordnung (EG) Nr. 111/2005 des Rates vom 22. Dezember 2004 zur Festlegung von Vorschriften für die Überwachung des Handels mit Drogenaustauschstoffen zwischen der Gemeinschaft und Drittländern. Online auf EUR-Lex, abgerufen am 31. Juli 2021.
- ↑ a b Verordnung des EDI über die Verzeichnisse der Betäubungsmittel, psychotropen Stoffe, Vorläuferstoffe und Hilfschemikalien. Online auf Fedlex, abgerufen am 1. August 2021.
- ↑ a b c d Verordnung des Bundesministeriums für Gesundheit vom 27. September 2022: Dritte Verordnung zur Änderung der Anlage des Neue-psychoaktive-Stoffe-Gesetzes. BGBl. Jg. 2022 Teil I Nr. 35, ausgegeben zu Bonn am 6. Oktober 2022, S. 1563.
- ↑ Deutscher Bundesrat: Plenarprotokoll (PDF) zur 1006. Sitzung am 25. Juni 2021, S. 284/338.
- ↑ Verena K. Angerer: Neue psychoaktive Substanzen in der forensischen Toxikologie: Synthetische Cannabinoide – Eine unendliche Geschichte? Dissertation, Albert-Ludwigs-Universität Freiburg im Breisgau 2017, S. 15. Online auf der Website der Universität Freiburg, abgerufen am 1. August 2021.
- ↑ Verordnung des Bundesministers für Gesundheit über Neue Psychoaktive Substanzen (Neue-Psychoaktive-Substanzen-Verordnung – NPSV). Online im Rechtsinformationssystem des Bundes, abgerufen am 31. Juli 2021.